Paeds · endocrinology-diabetes-and-growth
Short stature and poor linear growth
Also known as Short stature · Poor linear growth · Growth failure · Faltering linear growth
Fellowship guide to the short child: position, velocity and proportion drive the differential, from normal variants (familial short stature, constitutional delay) to GH deficiency, hypothyroidism, coeliac disease, syndromes, skeletal dysplasia and psychosocial causes, with a directed investigation pathway and cause-directed therapy including licensed growth-hormone indications.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Likely a variant
normal velocity
- Short parents
- Height velocity tracks a low parallel channel
- Normal examination and puberty
- Bone age consistent with age — reassure and follow
Likely pathological
poor velocity
- Downward centile crossing
- Disproportionate build or dysmorphism
- Systemic symptoms or weight gain with poor height
- Psychosocial red flags — investigate and refer
Short stature is defined by position; it is investigated because of velocity. Pair every cut-off with the serial trend and the family target. [2] [17]
Overview & Definition
Short stature is a descriptive auxological label, not a diagnosis. It means a child's height is more than two standard deviations below the mean for age and sex — roughly at or below the 3rd centile on a standard growth chart. About one child in forty sits there by definition, and most are healthy. [2] [5]
The clinical question is never "is this child short?" The question is "why is this child short, and is linear growth still on track?" A single low measurement answers the first part only. Serial measurements answer the second, and that is the part that matters. Growth velocity — the speed of change over time — separates the healthy short child from the child with a growth disorder. [2] [6]
Poor linear growth, or growth failure, is the loss of height potential: a child whose height centile falls across the chart, or whose velocity drops below the normal range for age. This may be the first sign of systemic disease, hormone deficiency, a genetic syndrome, undernutrition, or neglect. The fellowship task is to move from a low point on a chart to a defensible cause, using position, velocity, proportion and family context — then to choose observation, investigation or cause-directed therapy. [1] [6]
Classification
Start with the body, not the chart. Short stature divides by body proportion, because proportion changes the differential immediately. Proportionate short stature keeps a normal upper-to-lower body segment ratio for age. Disproportionate short stature does not, and points straight to a skeletal dysplasia or a defect of the SHOX gene. [2] [8]
Within proportionate short stature, the next split is velocity. Normal velocity with short parents is a normal variant — familial short stature, or constitutional delay of growth and puberty when the tempo is slow. Poor velocity, in contrast, means pathology somewhere in the growth engine: the endocrine system, the gut, the kidneys, a chronic illness, or the environment. [2] [5]
The short-child work-up in one line
6 Ps
Height at or below −2 SD for age and sex?
Is the height channel crossing down on serial plots?
Upper-to-lower segment and arm span — disproportionate?
Mid-parental (target) height corridor
Tanner stage and timing
Systemic, dysmorphic and psychosocial red flags
Work all six before you reach for a hormone test. [2] [6]
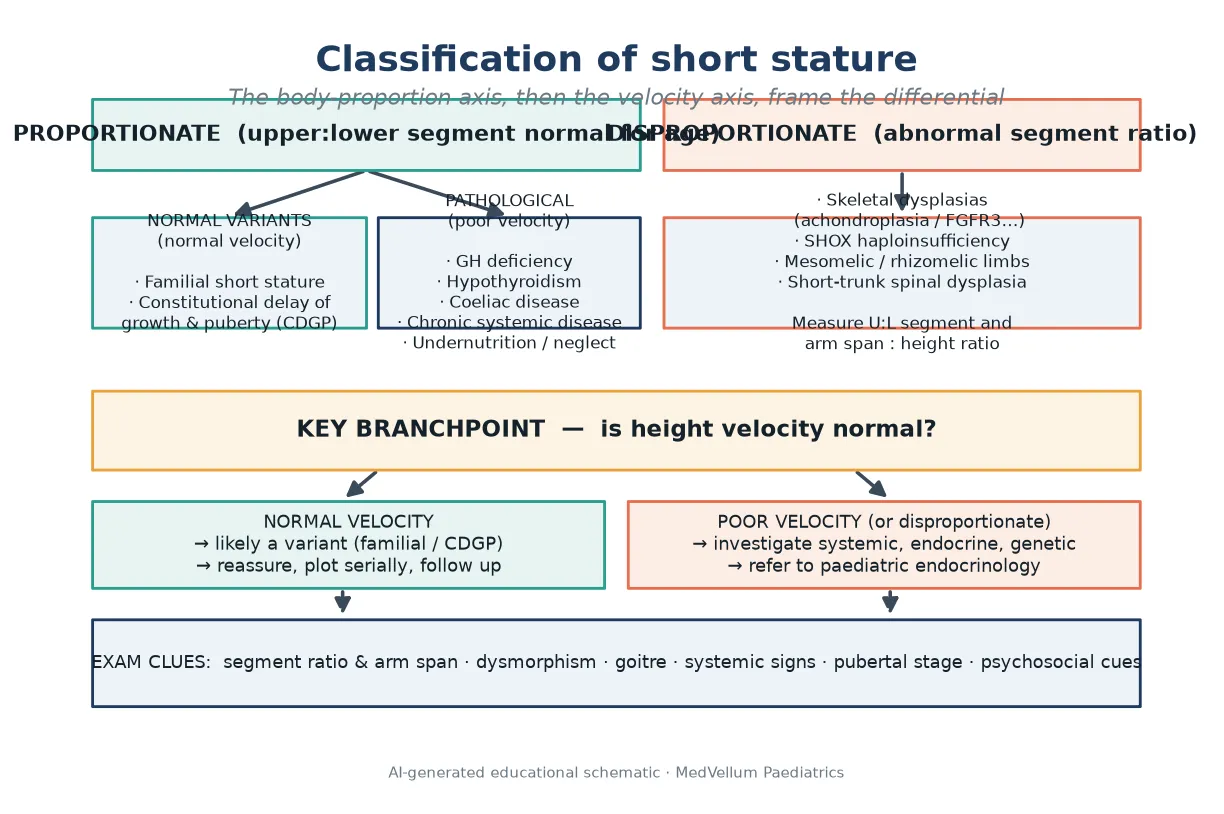
A third label arrives only at the end of the work-up. Idiopathic short stature is a residual diagnosis for a child whose height is below −2 SDS, who has been fully evaluated, and in whom no cause is found — including normal stimulated growth hormone testing under consensus criteria. It is never a first-glance clinic label for a small child. [1] [5]
Epidemiology & Risk Factors
By definition, about 2.3% of children sit below −2 SD on any growth chart, so short stature is common in primary care. The harder number is how many of those have treatable pathology. Among referred children, normal variants (familial short stature and constitutional delay) dominate; endocrine and systemic causes are individually rarer but carry the most to gain from detection. [2] [5]
Risk factors cluster by mechanism. Perinatal risk includes being born small for gestational age, prematurity, and poor catch-up growth. Systemic risk includes untreated coeliac disease, inflammatory bowel disease, chronic kidney disease, cyanotic congenital heart disease, and chronic anaemia. Endocrine risk covers growth hormone deficiency, hypothyroidism, glucocorticoid excess, and sex-steroid timing. Genetic risk includes Turner, Noonan and Prader-Willi syndromes, SHOX haploinsufficiency, and the skeletal dysplasias. [3] [8] [14]
Social risk is real and often missed. Food insecurity, neglect, and caregiver mental illness can produce psychosocial short stature with growth that recovers when the environment changes. Do not assign a biological cause to a child whose growth normalises away from the index household. [16]
Pathophysiology
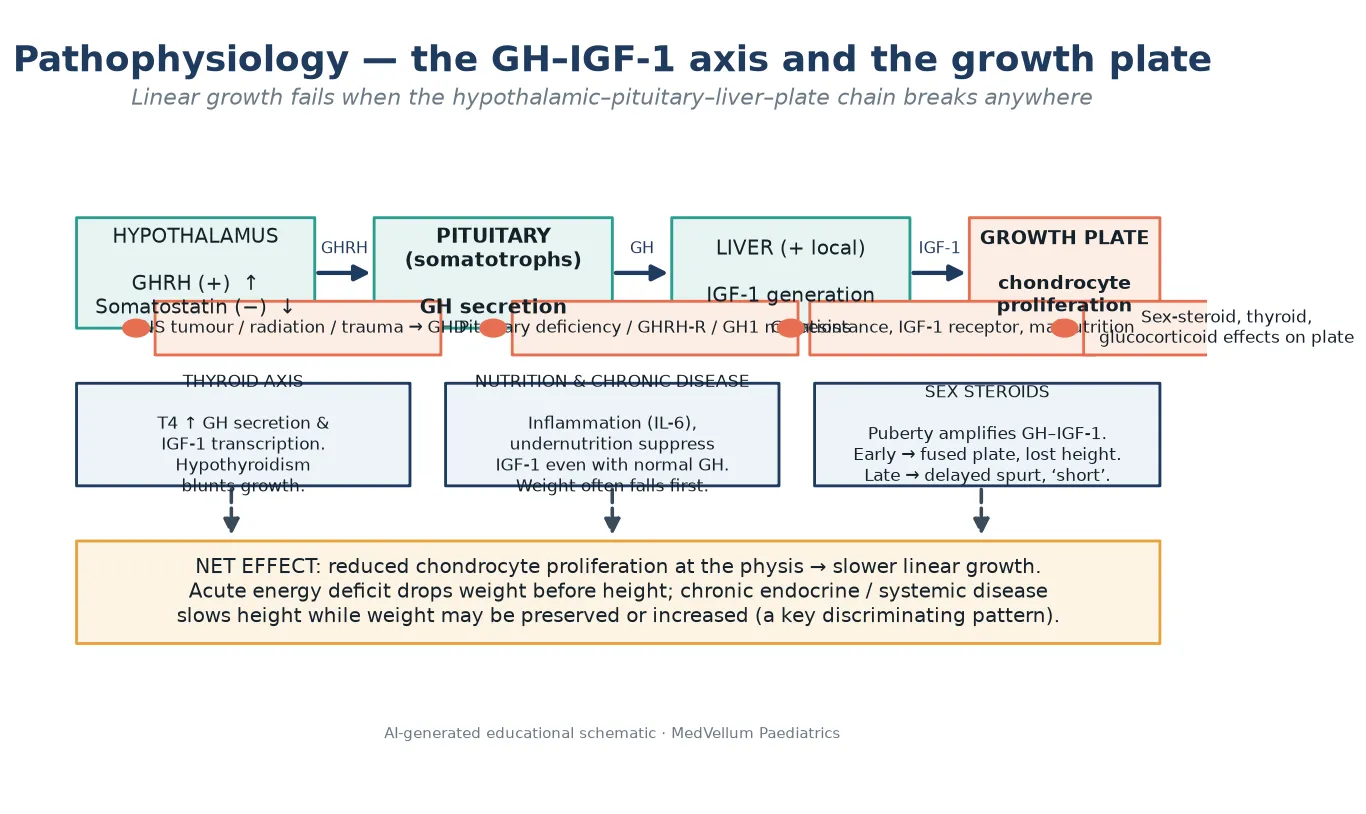
Linear growth is the net output of a chain that runs from the hypothalamus to the growth plate. Each link can fail, and the failure pattern predicts the cause. [4] [7]
The hypothalamus releases growth-hormone-releasing hormone to stimulate, and somatostatin to inhibit, the pituitary somatotrophs. The pituitary secretes growth hormone, which acts on the liver and locally at the growth plate to generate insulin-like growth factor 1 (IGF-1). IGF-1 drives chondrocyte proliferation in the physis — the cartilage engine that lengthens bone. Break the chain at the brain (tumour, radiation, trauma), the pituitary (genetic or structural deficiency), the liver (malnutrition, chronic inflammation), or the plate (sex-steroid or glucocorticoid effects), and linear growth slows. [4] [6]
Three other axes modify the chain. Thyroid hormone is required for both growth hormone secretion and IGF-1 action, so acquired hypothyroidism blunts growth. Nutrition and chronic inflammation suppress IGF-1 generation even when growth hormone is normal — which is why systemic disease and undernutrition slow height while weight often falls first. Sex steroids at puberty amplify growth hormone action and produce the growth spurt; arrive too early and they fuse the growth plate and steal adult height, arrive late and they delay the spurt so the child looks short for longer. [7] [14]
A useful clinical rule follows from this physiology. Acute energy deficit drops weight before height. Chronic endocrine disease — hypothyroidism, growth hormone deficiency, Cushing syndrome — often slows height while weight is preserved or rising. That weight-versus-height pattern is a discriminator you can read off the chart. [2] [6]
Clinical Presentation
Short stature most often presents as a concern raised by parents, a teacher, or a primary-care growth check: "she is the smallest in the class," "his clothes still fit from last year," "the school nurse flagged his height." The presentation is usually incidental rather than symptomatic, which is why accurate serial measurement is the gateway. [2]
The pattern of presentation points to the cause. A child who has always tracked a low parallel channel, with short parents, presents as a healthy variant. A child whose growth has decelerated — crossing centiles downward on serial plots — presents with possible pathology. A child with a disproportionate build, short limbs, or a short trunk suggests a skeletal dysplasia or SHOX deficiency. [2] [8]
Atypical and syndromic presentations matter. A short girl with a webbed neck, broad chest and delayed puberty may have Turner syndrome, which can be missed because the phenotype is sometimes subtle. A short child with feeding difficulty, hypotonia and developmental concern may have Prader-Willi syndrome in early life. A short child with headache, visual change or a new squint may harbour a pituitary or hypothalamic tumour compressing the growth-hormone axis. [4] [9] [15]
The adolescent presents through the lens of puberty timing. Constitutional delay of growth and puberty looks like short stature with a late spurt; precocious puberty looks tall now but threatens final height. Ask about body image, eating, and growth anxiety, and open confidential enquiry where the young person is able to consent. [2] [6]
Differential Diagnosis
Familial short stature
normal velocity, parallel channel
- Short parents
- Height tracks a low channel parallel to the centiles
- Bone age matches chronological age
- Normal exam and puberty — reassure and follow
Constitutional delay of growth and puberty
slow tempo
- Delayed growth tempo
- Delayed bone age
- Late but eventually complete puberty
- Often a family history of late bloomers — final height usually in the family range
The pathological differentials fall into groups that each change the investigation and the plan. [2] [6]
Endocrine. Growth hormone deficiency presents with slow linear growth, a cherubic or immature appearance, and preserved or increased weight; IGF-1 and IGFBP-3 are low, and stimulation testing is abnormal. Acquired hypothyroidism slows growth, often with weight gain, fatigue, constipation and goitre. Cushing syndrome and glucocorticoid exposure suppress growth while weight rises. [6] [14]
Gastrointestinal and systemic. Coeliac disease can present as isolated short stature before any gut symptoms, which is why serology screens short children in many pathways. Inflammatory bowel disease, chronic kidney disease, cyanotic congenital heart disease, chronic anaemia and poorly controlled asthma all consume the growth engine through inflammation, poor intake, or treatment effects. [2] [6]
Genetic and syndromic. Turner syndrome (45,X and variants) carries short stature, ovarian failure and a phenotype that may be subtle in mosaics — karyotype is diagnostic. Noonan syndrome mimics Turner in a male or female child with a RASopathy phenotype and often congenital heart disease. Prader-Willi syndrome presents with infantile hypotonia, feeding difficulty and later hyperphagia with short stature. SHOX haploinsufficiency causes short stature with Madelung deformity and mesomelic shortening, sometimes without obvious disproportion. [8] [9] [10] [15]
Disproportionate — skeletal dysplasia. Achondroplasia and the FGFR3 family produce rhizomelic short limbs with a relatively large head and normal trunk length. Other dysplasias shorten the trunk or the forearms and lower legs. An abnormal upper-to-lower segment ratio or arm-span-to-height mismatch is the bedside signal to image and refer. [2] [8]
Perinatal. Small-for-gestational-age children are expected to catch up; those who do not by two to four years are a defined group with their own growth-hormone pathway. [3]
Psychosocial. Psychosocial short stature (deprivation dwarfism) presents with poor growth in an environment of neglect or abuse, often with a history that does not fit, and growth that recovers when the child is removed to a safe setting. Growth recovery lines on imaging may mark the recovery after deprivation. [16]
Clinical & Bedside Assessment
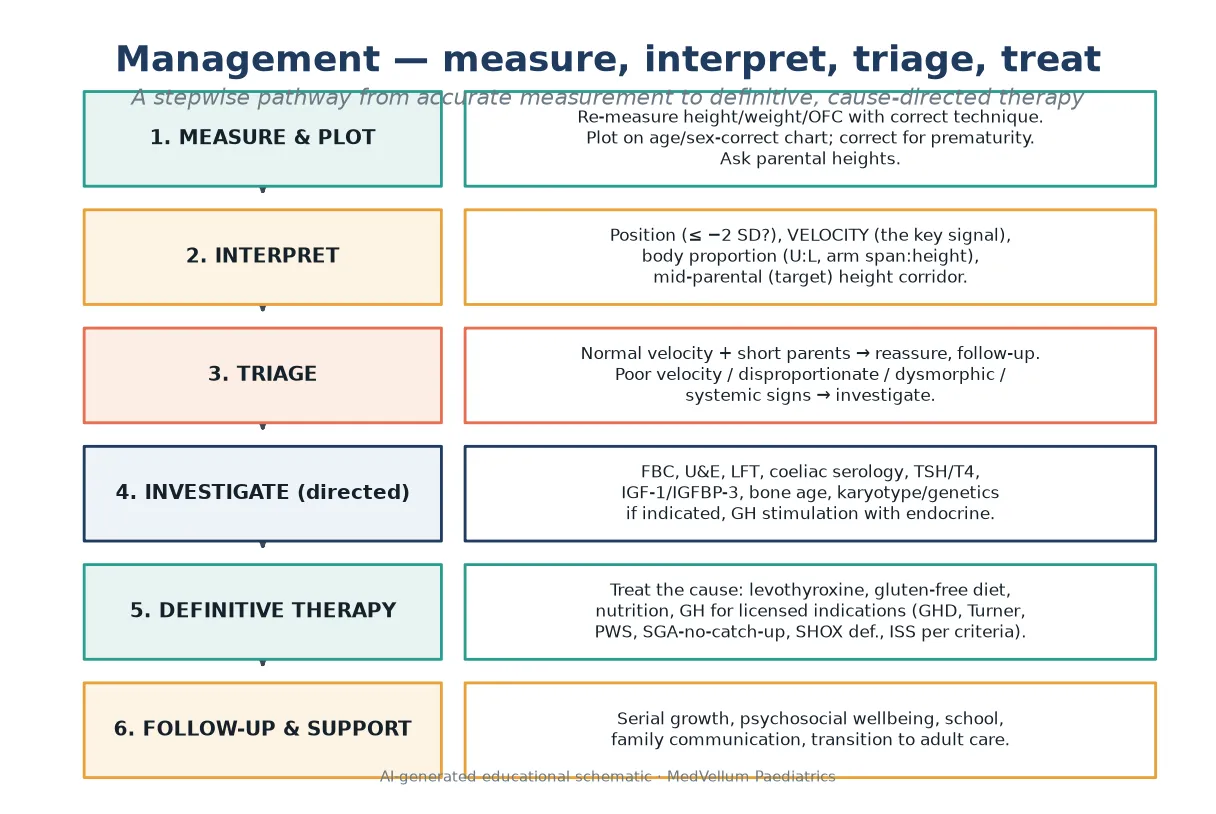
Measurement is the first investigation, and most "short stature" referrals have a measurement problem. Re-measure the child yourself before you interpret. For children under two, take supine length with two operators, naked or dry-nappy weight, and occipitofrontal circumference. From about two years, measure standing height with heels, buttocks and shoulders against the stadiometer, shoes off, head in the Frankfort plane. Plot immediately, on the chart for the correct age and sex, and correct for prematurity in early childhood. [2]
Assess body proportion at the bedside. The upper-to-lower segment ratio falls from about 1.7 at birth to near 1.0 by mid-childhood; a ratio that is wrong for age, or an arm span that differs from height by more than about 5 cm, flags disproportionate short stature and points toward skeletal dysplasia or SHOX deficiency. Measure and document it rather than guess. [2] [8]
Calculate the mid-parental (target) height to frame the genetic corridor. Using the Tanner approach, the target for a boy is the average of parental heights plus 6.5 cm, and for a girl the average minus 6.5 cm, with a corridor of roughly ±10 cm (two standard deviations) around it. Treat the result as a corridor, not a promise — recent work shows the formula is imprecise, especially when parents are at the extremes — but a child whose height is far below the family target is a child who needs explaining. [2] [17]
What is the Tanner mid-parental height formula?
For a boy: (father's height + mother's height) / 2 + 6.5 cm. For a girl: (father's height + mother's height) / 2 − 6.5 cm. Expect the child to track within about ±10 cm of this. A height far below the target corridor warrants investigation even if the current centile looks "normal" for the population. [17]
Stage puberty (Tanner) when you interpret an adolescent, because pubertal timing changes both the velocity and the expected final height. Then complete a whole-child examination: look for dysmorphism, goitre, signs of chronic disease, abdominal masses, skin striae, and nutritional status; and screen deliberately for safeguarding and psychosocial red flags. [2] [16]
Investigations
Many short children need no panel at all. A healthy variant with short parents, normal velocity, a normal exam and a bone age consistent with age is observed, not investigated. Reserve testing for poor velocity, disproportionate build, dysmorphism, systemic symptoms, a height far below the family target, or psychosocial concern. [1] [6]
Use a directed first-line panel for the child who warrants investigation: full blood count, electrolytes and renal function, liver function, bone profile, coeliac serology in any gluten-exposed child, inflammatory markers where indicated, and thyroid function (TSH and free T4). Add IGF-1 and IGFBP-3 as screening for the growth-hormone axis — low values support deficiency, normal values do not exclude it. A bone age radiograph assesses growth tempo and remaining height potential but does not by itself diagnose growth-hormone deficiency. [2] [6]
Second-line and specialist testing depends on the pattern. Karyotype or chromosomal microarray is indicated for any short girl in whom Turner is possible, and for dysmorphic or syndromic children. Growth-hormone stimulation testing belongs to paediatric endocrinology, is reserved for children with a credible pre-test probability of deficiency, and uses pharmacological stimuli after priming where local protocols require — because random growth hormone levels are pulsatile and unhelpful. Cranial imaging follows when a central cause is suspected or confirmed, to look for a pituitary or hypothalamic lesion. [4] [6]
Management — Resuscitation
Most short stature is not an emergency, but three situations outrank the chart debate. Severe acquired hypothyroidism with decompensation needs urgent endocrine stabilisation, not a slow outpatient work-up. A short child with headache, visual change, papilloedema or new cranial nerve signs may have a pituitary or hypothalamic tumour causing growth-hormone deficiency and raised intracranial pressure — image and escalate immediately. [4] [14]
Severe undernutrition, dehydration, hypothermia or sepsis from neglect needs acute medical care first, with safeguarding escalation in parallel. Psychosocial short stature with an unsafe environment is a child-protection emergency as much as a growth problem; stabilise the child, document objectively, and follow local safeguarding pathways without delay. [16]
Management — Definitive & Stepwise
Definitive management is cause-directed. Treat the disease, and the growth usually follows. [6]
- Confirm the measurement, the chart, and the velocity before any intervention.
- Reassure and observe the normal variant — familial short stature and constitutional delay need explanation, serial plotting, and follow-up, not hormones.
- Treat the identified cause: levothyroxine for hypothyroidism, a gluten-free diet for coeliac disease, nutrition and disease control for chronic illness, and a safe environment for psychosocial short stature.
- Refer to paediatric endocrinology for the child who may need growth-hormone therapy. [2] [6]
Growth-hormone therapy is licensed for specific indications and is not a height booster for every short child. Recombinant human growth hormone is given as a daily subcutaneous injection, commonly at about 0.18 to 0.30 mg/kg/week in divided daily doses for growth-hormone deficiency, titrated to the growth response and IGF-1 level. Licensed indications include proven growth-hormone deficiency, Turner syndrome, Prader-Willi syndrome, short children born small for gestational age who fail to catch up, SHOX deficiency, chronic kidney disease–related growth failure in some jurisdictions, and idiopathic short stature that meets strict criteria. [1] [4] [13]
The benefit in idiopathic short stature is modest and is weighed against years of daily injections, cost, and a small adverse-effect burden — shared decision-making is essential, not optional. Recombinant IGF-1 (mecasermin) is reserved for the rare child with severe primary IGF-1 deficiency or growth-hormone receptor resistance who cannot respond to growth hormone. [11] [18]
Follow-up is part of the treatment. Set explicit review intervals, plot serially, monitor psychosocial wellbeing and school function, communicate plainly with the family, and plan transition to adult endocrinology for adolescents with ongoing growth or hormone needs. [3] [10]
Specific Subtypes & Scenarios
Familial short stature. Short parents, a normal exam, normal velocity, and a bone age near chronological age. Reassure, plot serially, and do not investigate or treat. [2]
Constitutional delay of growth and puberty. Slow tempo, delayed bone age, late puberty, often a family history of late bloomers. Growth eventually catches up into the family range. Counselling and follow-up are the management; sex steroids are occasionally used to induce puberty under endocrine guidance. [2] [6]
Growth-hormone deficiency. Slow linear growth with preserved weight, immature appearance, low IGF-1/IGFBP-3, and an abnormal stimulation test. Magnetic resonance imaging looks for a structural pituitary cause. Growth-hormone replacement restores velocity; the response is best when treatment starts early. [4] [7]
Turner syndrome. Short stature is near-universal and is driven partly by SHOX haploinsufficiency. Growth hormone improves height and is a licensed indication; karyotype confirms the diagnosis, and cardiac and ovarian surveillance run alongside growth management. [8] [9]
Noonan syndrome. A RASopathy with short stature, often congenital heart disease and a distinctive face. Growth hormone is used in many jurisdictions; the response is variable and cardiac status guides the plan. [10]
Prader-Willi syndrome. Infantile hypotonia and feeding failure give way to hyperphagia, obesity and short stature with hypogonadism. Growth hormone improves body composition, linear growth and perhaps cognition, with strict sleep-apnoea screening before and during treatment. [15]
SHOX deficiency and skeletal dysplasia. SHOX haploinsufficiency causes short stature with mesomelic shortening and Madelung deformity; skeletal dysplasias such as achondroplasia are disproportionate. The body-proportion exam is the gateway, and growth hormone or targeted therapies depend on the specific diagnosis. [2] [8]
Small-for-gestational-age with no catch-up. Children who do not catch up by two to four years are a defined group for whom growth hormone is licensed under consensus criteria, with attention to metabolic risk in later life. [3]
Coeliac disease and systemic causes. A gluten-free diet restores growth in coeliac-related short stature; control of chronic kidney disease, inflammatory bowel disease, or anaemia lifts the brake on growth where the underlying disease allows. [2]
Psychosocial short stature. Growth recovers when the child is moved to a safe, nurturing environment. Document objectively, escalate safeguarding, and avoid labelling the family in front of the child. [16]
Complications & Pitfalls
The commonest error is diagnosing disease from a single low point without velocity. A second is using the wrong chart or the wrong sex, or failing to correct for prematurity, so a healthy child is labelled pathological. A third is over-investigating the familial variant with short parents and normal velocity, or — the opposite trap — under-investigating the "well-looking" child whose centile is quietly falling. [2] [6]
Stigmatising language about height or size loses the family and the follow-up. Promising adult height from the mid-parental formula alone ignores its imprecision. Missing safeguarding while debating centiles, or missing a pituitary tumour behind a "constitutional delay" label, are the serious misses. Growth-hormone therapy given outside licensed indications wastes cost and carries avoidable risk; giving it without shared decision-making erodes trust. [4] [16] [17]
Prognosis & Disposition
A normal variant reaches an adult height in the family range; constitutional delay catches up late. The prognosis of pathological short stature depends on the cause and on how early treatment starts — growth-hormone deficiency corrected early restores most height potential, while chronic disease left unchecked permanently loses centimetres. [6] [7]
Disposition spans primary care for reassured variants, general paediatric follow-up for the child under observation, paediatric endocrinology for the child on a growth-hormone pathway, and multidisciplinary involvement for syndromic, renal, gastrointestinal and psychosocial causes. Transition to adult care matters for adolescents with continuing endocrine, renal or metabolic needs. [3] [10]
Special Populations
Children with syndromic and complex chronic disease need individual growth baselines and condition-specific charts where validated, alongside their other surveillance. Small-for-gestational-age and ex-preterm graduates need catch-up surveillance and metabolic-risk counselling. Adolescents need confidential, body-image-aware enquiry and a planned transition. [3] [10]
Indigenous families need culturally safe conversations about growth without racialised chart misuse. Migrant and refugee children may arrive with interrupted records and dual infection-and-nutrition risk. Out-of-home-care growth is both a welfare and a medical indicator. Children with disability need adapted measurement and interpretation. In every case, plot, document, and read the trend rather than a single number. [2] [16]
Evidence, Guidelines & Regional Differences
The Growth Hormone Research Society consensus sets the framework for diagnosing and treating growth-hormone deficiency in childhood and adolescence. [4] The 2008 consensus on idiopathic short stature defines it as a residual label after complete evaluation, including stimulated growth-hormone testing, and frames who might be considered for therapy. [1] Wit and colleagues set out the definition, epidemiology and diagnostic evaluation of idiopathic short stature. [5]
Murray and colleagues summarise the controversies in diagnosing growth-hormone deficiency, including the limits of stimulation testing. [6] Boguszewski and colleagues review growth-hormone deficiency and replacement in children. [7] Cohen and colleagues report the dose-sparing and safety profile of an IGF-1-based growth-hormone dosing regimen. [13] Deodati and colleagues review the randomised trials of growth hormone in idiopathic short stature, and Paltoglou and colleagues report a meta-analysis of its effect on adult height. [12] [18]
The 2023 international consensus guideline on small-for-gestational-age covers etiology and management from infancy to early adulthood, including growth-hormone eligibility. [3] The congenital hypothyroidism 2020–2021 consensus update covers the thyroid cause of poor growth and its treatment. [14] SHOX haploinsufficiency diagnosis and treatment is reviewed by Jorge and colleagues. [8] Baxter and colleagues (Cochrane) summarise growth hormone in Turner syndrome. [9] Dahlgren and Noordam review Noonan syndrome growth and treatment. [10] Cassidy and colleagues review Prader-Willi syndrome. [15] Hiraoka and colleagues compare growth recovery lines in psychosocial versus other short stature. [16] Ciancia and colleagues examine how accurate Tanner's target-height formula really is. [17]
Exam Pearls
- Velocity beats position: a falling height centile is pathology until proven otherwise. [2] [6]
- Proportion first: an abnormal upper-to-lower segment ratio or arm span sends you to skeletal dysplasia or SHOX deficiency. [8]
- Idiopathic short stature is residual after full evaluation including stimulated growth-hormone testing — never a first-glance label. [1] [5]
- Weight rising while height falls points to endocrine disease (hypothyroidism, Cushing, growth-hormone deficiency). [2] [6]
- Small for gestational age is size at birth; fetal growth restriction is a fetal process — not every SGA child had FGR, and not every FGR fetus is still SGA at birth. [3]
- Short stature plus headache or visual change: think pituitary or hypothalamic tumour — image, do not reassure. [4]
- Growth hormone is a daily subcutaneous injection at about 0.18 to 0.30 mg/kg/week, licensed for specific indications, not a height booster for every short child. [1] [13]
- The mid-parental height formula gives a corridor of ±10 cm, not a promise; it is imprecise at the extremes. [17]
Definition & signal
- Height ≤ −2 SD for age/sex (≈ 3rd centile)
- Growth velocity — crossing down = pathology
- Proportion: upper:lower segment, arm span:height
Family & variants
- Mid-parental target ± 10 cm corridor
- Variant triad: familial short stature · constitutional delay · normal velocity
Causes
- Endocrine: GH deficiency · hypothyroidism · Cushing/glucocorticoids
- Disproportionate: skeletal dysplasia · SHOX deficiency
- Syndromic: Turner · Noonan · Prader-Willi
- Systemic: coeliac · IBD · CKD · congenital heart disease
Therapy
- Treat the cause: levothyroxine · gluten-free diet · nutrition · safe environment
- GH ≈ 0.18–0.30 mg/kg/week subcut, licensed indications only
Every cut-off above must be read with the velocity trend, the proportion and the family target. [2] [6] [17]
References
- [1]Cohen P, Rogol AD, Deal CL, et al Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. The Journal of Clinical Endocrinology and Metabolism, 2008.PMID 18782877
- [2]Barstow C, Rerucha C Evaluation of Short and Tall Stature in Children. American Family Physician, 2015.PMID 26132126
- [3]Hokken-Koelega ACS, van der Steen M, Boguszewski MCS, et al International Consensus Guideline on Small for Gestational Age: Etiology and Management From Infancy to Early Adulthood. Endocrine Reviews, 2023.PMID 36635911
- [4]Growth Hormone Research Society Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. The Journal of Clinical Endocrinology and Metabolism, 2000.PMID 11095419
- [5]Wit JM, Clayton PE, Rogol AD, et al Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth Hormone & IGF Research, 2008.PMID 18182313
- [6]Murray PG, Dattani MT, Clayton PE Controversies in the diagnosis and management of growth hormone deficiency in childhood and adolescence. Archives of Disease in Childhood, 2016.PMID 26153506
- [7]Boguszewski MCS, Cardoso-Demartini AA, Boguszewski CL, et al Growth hormone deficiency and replacement in children. Reviews in Endocrine & Metabolic Disorders, 2021.PMID 33029711
- [8]Jorge AA, Funari MF, Nishi MY, et al Short stature caused by isolated SHOX gene haploinsufficiency: update on the diagnosis and treatment. Pediatric Endocrinology Reviews, 2010.PMID 21150837
- [9]Baxter L, Bryant J, Cave CB, Milne R Recombinant growth hormone for children and adolescents with Turner syndrome. Cochrane Database of Systematic Reviews, 2007.PMID 17253498
- [10]Dahlgren J, Noordam C Growth, Endocrine Features, and Growth Hormone Treatment in Noonan Syndrome. Journal of Clinical Medicine, 2022.PMID 35407641
- [11]Collett-Solberg PF, Misra M; Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society The role of recombinant human insulin-like growth factor-I in treating children with short stature. The Journal of Clinical Endocrinology and Metabolism, 2008.PMID 18165284
- [12]Deodati A, Peschiaroli E, Cianfarani S Review of growth hormone randomized controlled trials in children with idiopathic short stature. Hormone Research in Paediatrics, 2011.PMID 21912163
- [13]Cohen P, Weng W, Rogol AD, et al Dose-sparing and safety-enhancing effects of an IGF-I-based dosing regimen in short children treated with growth hormone in a 2-year randomized controlled trial: therapeutic and pharmacoeconomic considerations. Clinical Endocrinology, 2014.PMID 24428305
- [14]van Trotsenburg P, Stoupa A, Kassis M, et al Congenital Hypothyroidism: A 2020-2021 Consensus Guidelines Update-An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid, 2021.PMID 33272083
- [15]Cassidy SB, Schwartz S, Miller JL, Driscoll DJ Prader-Willi syndrome. Genetics in Medicine, 2012.PMID 22237428
- [16]Hiraoka T, Nishizaki M, Saito T, et al Growth recovery lines in children: A comparison between psychosocial short stature and other pathological causes of short stature. Child Abuse & Neglect, 2022.PMID 34801849
- [17]Ciancia S, Cajas PR, Binot J, et al How accurate is Tanner's formula in estimating target height? BMC Pediatrics, 2025.PMID 41350675
- [18]Paltoglou G, Dimitropoulos I, Vlachopapadopoulou E The effect of treatment with recombinant human growth hormone (rhGH) on linear growth and adult height in children with idiopathic short stature (ISS): a systematic review and meta-analysis. Journal of Pediatric Endocrinology and Metabolism, 2020.PMID 33035189