Paeds · genetics-dysmorphology-and-metabolism
Dysmorphology examination and syndrome recognition
Also known as Dysmorphology examination · Dysmorphic features assessment · Congenital anomaly examination · Syndrome recognition · Clinical dysmorphology
Fellowship approach to a structured dysmorphology examination using standard Elements of morphology terminology, classifying anomalies by mechanism and significance, generating and narrowing a syndrome differential, ordering first-tier chromosomal microarray then exome or genome, and communicating uncertainty and recurrence risk to a family.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
D.Y.S.M.O.R.P.H. exam
Overview & Definition
Dysmorphology is the clinical discipline of detecting, describing and interpreting structural variations, then using the pattern to recognise an underlying syndrome. It was built on a simple, powerful idea: a single odd feature is usually harmless, but a constellation of features points to a shared cause. The examination itself is therefore an exercise in disciplined description before diagnosis. You are collecting evidence, not jumping to a label. [1]
The examination differs from a general physical in three ways. First, it uses a controlled vocabulary — the Elements of morphology project — so that a clinician in Auckland and one in Aberdeen describe the same face in the same words. Second, it relies on measurements compared with population norms, because the eye is poor at judging whether a feature is truly abnormal or merely familial. Third, it deliberately looks for a pattern across body regions, since the diagnostic value lives in the combination rather than in any one sign. [1] [5]
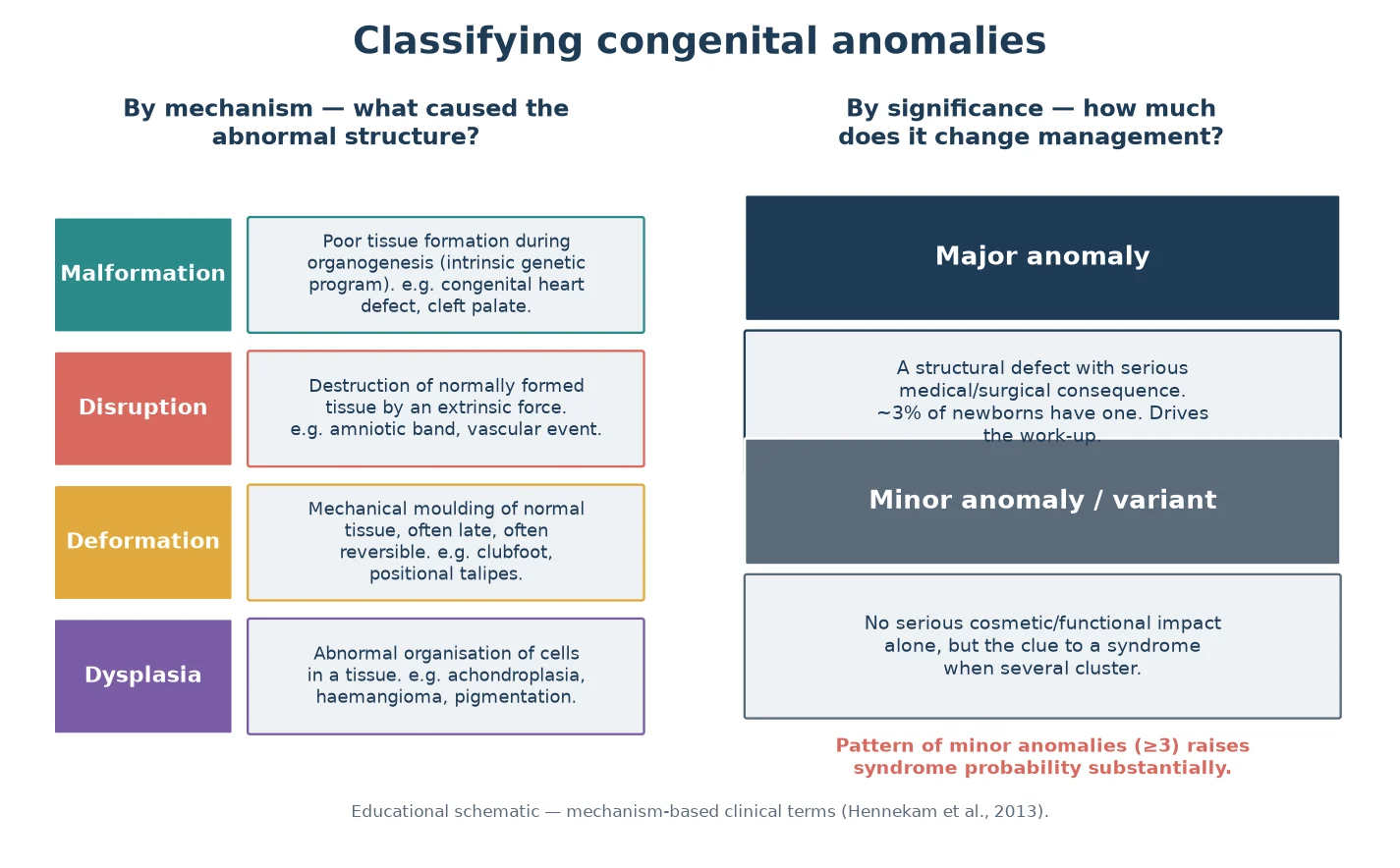
A practical distinction runs through the whole assessment: a major anomaly has a real structural, medical or surgical consequence (a heart defect, cleft palate, neural tube defect), whereas a minor anomaly or normal variant does not, on its own, change management. The trap is to dismiss minor anomalies as cosmetic. Three or more minor anomalies in the same child substantially raises the chance of an underlying syndrome, because each one slightly shifts the prior probability and together they demand a search. [3] [5]
Classification
Classify every anomaly along two axes and you will already have begun the differential. The first axis is mechanism: a malformation is poor tissue formation during organogenesis, a disruption is destruction of normally formed tissue by an extrinsic force, a deformation is mechanical moulding of normal tissue (often late and often reversible), and a dysplasia is abnormal organisation of cells within a tissue. A cleft palate and a congenital heart defect are malformations; an amniotic-band limb defect is a disruption; a positional clubfoot is a deformation; achondroplasia is a dysplasia. [3] [5]
The second axis is significance: major versus minor, as above. Holding both axes in mind lets you reason quickly. A single major malformation such as an isolated cardiac defect is common and often multifactorial; but pair it with even one suggestive facial feature and you must widen the search. Mechanism also predicts reversibility and prognosis — a deformation may resolve once the mechanical force is removed, while a malformation is fixed. [3]
Beyond individual anomalies, classify the overall pattern. A syndrome is a recognised set of anomalies with a shared cause (Down syndrome). An association is a non-random co-occurrence without a single defined cause (VACTERL). A sequence is a cascade where one initial anomaly produces the rest (Robin sequence: micrognathia drives glossoptosis which drives cleft palate). A field defect is abnormal development of a single embryonic region (the cardiac field, the limb field). Naming the pattern type sharpens both the differential and the counselling. [1] [3]
Epidemiology & Risk Factors
Major congenital anomalies are common: roughly two to four in every hundred liveborn infants has one, and congenital anomalies are a leading contributor to infant mortality and childhood disability worldwide. Most individual anomalies are multifactorial, arising from an interplay of genetic susceptibility and environment, which is why a careful pregnancy and family history is part of the examination rather than an optional extra. [3] [7]
Several factors lower the threshold for a detailed dysmorphology assessment. Advanced maternal age raises the chance of chromosomal aneuploidy; advanced paternal age raises the chance of certain new dominant mutations. A known teratogen exposure — alcohol, anticonvulsants, isotretinoin, warfarin, maternal diabetes or infection — points the examination toward a characteristic pattern. Consanguinity, a previously affected child, or a family history of miscarriage or stillbirth all raise the prior probability of a monogenic or recessive syndrome. [13] [7]
Pathophysiology
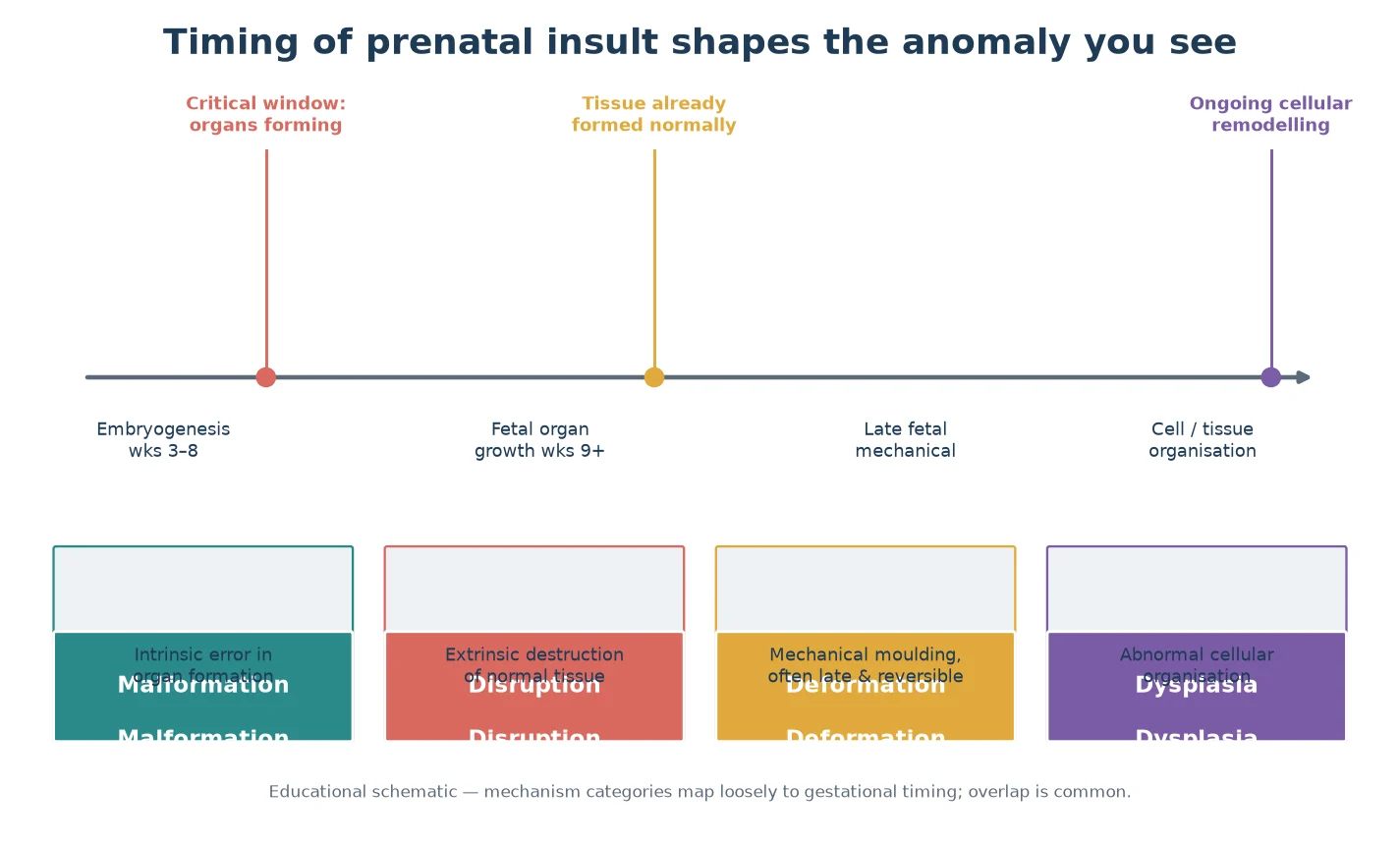
The type of anomaly you see is shaped by when in development the disruption struck. During early embryogenesis, when organs are being formed, an intrinsic error in the developmental program produces a malformation. Later, when tissues are already formed, a mechanical force moulds normal tissue into a deformation, while a destructive event — vascular, infective, or mechanical — produces a disruption. Ongoing cellular disorganisation produces a dysplasia, which may declare itself after birth. Timing is why a single insult can produce different anomalies in different organs, and why the same genetic change can look different from one child to the next. [1] [3]
This matters clinically in two ways. First, mechanism and timing predict what else to look for: a child with a disruption in one region should still be examined for malformations elsewhere, because insults can co-occur. Second, they shape prognosis and counselling — a deformation may improve once the cause is removed, whereas a malformation is fixed and a dysplasia may progress. Holding the developmental cascade in mind stops the examiner from over-focusing on the most visible feature and missing the pattern. [3]
Clinical Presentation
Children reach a dysmorphology assessment through one of several doors. A newborn may be flagged in the delivery room or nursery for an unusual face or an obvious major anomaly. An infant may be referred because development is lagging and the face looks different. An older child may arrive because a teacher or a relative commented on appearance or learning. Sometimes the trigger is a known family syndrome and the question is whether the child is affected. Each door sets a different tempo for the assessment. [7]
What you look for is anchored on high-yield regions. On the face, inspect head shape and size, the forehead, the eyes and orbits, the nasal bridge and tip, the philtrum and upper lip, the mouth and palate, the ears, and the neck. On the body, examine the hands and feet (digits, palmar creases, nail beds), the chest and abdomen, the spine, and the skin. Measuring matters as much as looking: occipitofrontal circumference, and the inner canthal, interpupillary and palpebral fissure distances, ear length, and philtrum and lip measurements, all judged against published norms. [1] [2]
Some patterns point straight to a teratogen. The fetal alcohol spectrum facies — a thin smooth philtrum, a thin vermilion border of the upper lip, and small palpebral fissures — is a classic example of a recognisable, exposure-related gestalt, and demonstrating how such a pattern is judged (measurement against norms plus a corroborating exposure history) is a high-yield viva skill. [13]
Differential Diagnosis
Generating the differential is a three-step move. First, strip the features down to those that are truly abnormal rather than familial or ethnic, using measurements against the correct population norms. Second, group the abnormal findings into a pattern and compare that pattern against recognised syndromes using databases such as the London Dysmorphology Database, OMIM and the Human Phenotype Ontology, or a deep-learning facial-analysis tool as a support. Third, narrow using the family history, pregnancy history and the tempo of presentation. [5] [12]
The can't-miss distinction is between a normal variant and a pathologic feature. A child whose parents share a particular nose or ear shape usually has a familial, not a syndromic, finding; measuring the parents settles much of this. Equally important is separating a sequence from its underlying syndromic cause. Robin sequence — micrognathia, glossoptosis and cleft palate — can occur in isolation, but it is also the presenting pattern of Stickler syndrome and other conditions, so the isolated label is never accepted until the syndromic causes are excluded. [3] [5]
Objective tools have changed how the differential is narrowed. Three-dimensional facial imaging and deep-learning systems can match a facial gestalt to known syndromes with high accuracy in validation studies, and they genuinely help when the clinician has not seen the rare pattern before. They are a support for, never a replacement for, structured clinical examination and genetic confirmation. [6] [12]
Clinical & Bedside Assessment
Observation before touch
Begin by watching the undressed child from across the room, ideally in good lighting and with the parent holding the child to settle them. Note the overall gestalt, the head shape, the proportions, and how the child moves and interacts, because the first impression often directs the systematic examination that follows. Observation is deliberately done before instruments and hands intervene, while the child is calm. [1] [2]
Systematic head-to-toe examination
Move in a fixed order — skull and face, then neck and chest, then abdomen and genitalia, then spine, limbs, skin and neurology — so that no region is missed. Describe each finding in standard terminology: prefer hypotelorism or hypertelorism, upslanting or downslanting palpebral fissures, a smooth philtrum, a thin vermilion border, low-set or posteriorly rotated ears, rather than vague phrases such as funny-looking. Vague language fails the examination and fails the record. [1] [2]
Measurements and photography
Measure the occipitofrontal circumference and the key facial distances, and plot them where centiles exist. Standardised clinical photographs — frontal, lateral and a view of any specific feature — become part of the longitudinal record and allow comparison over time and between relatives. Photography is performed with consent and stored according to local policy. [1] [6]
Examine the family
When a familial or syndromic cause is suspected, examine the parents and relevant siblings, because a parent with subtle features of the same condition changes both the diagnosis and the recurrence risk. This is one of the most under-used and highest-yield steps in the assessment. [5] [7]
Yield of tiered genetic testing in unexplained developmental delay or congenital anomalies
Investigations
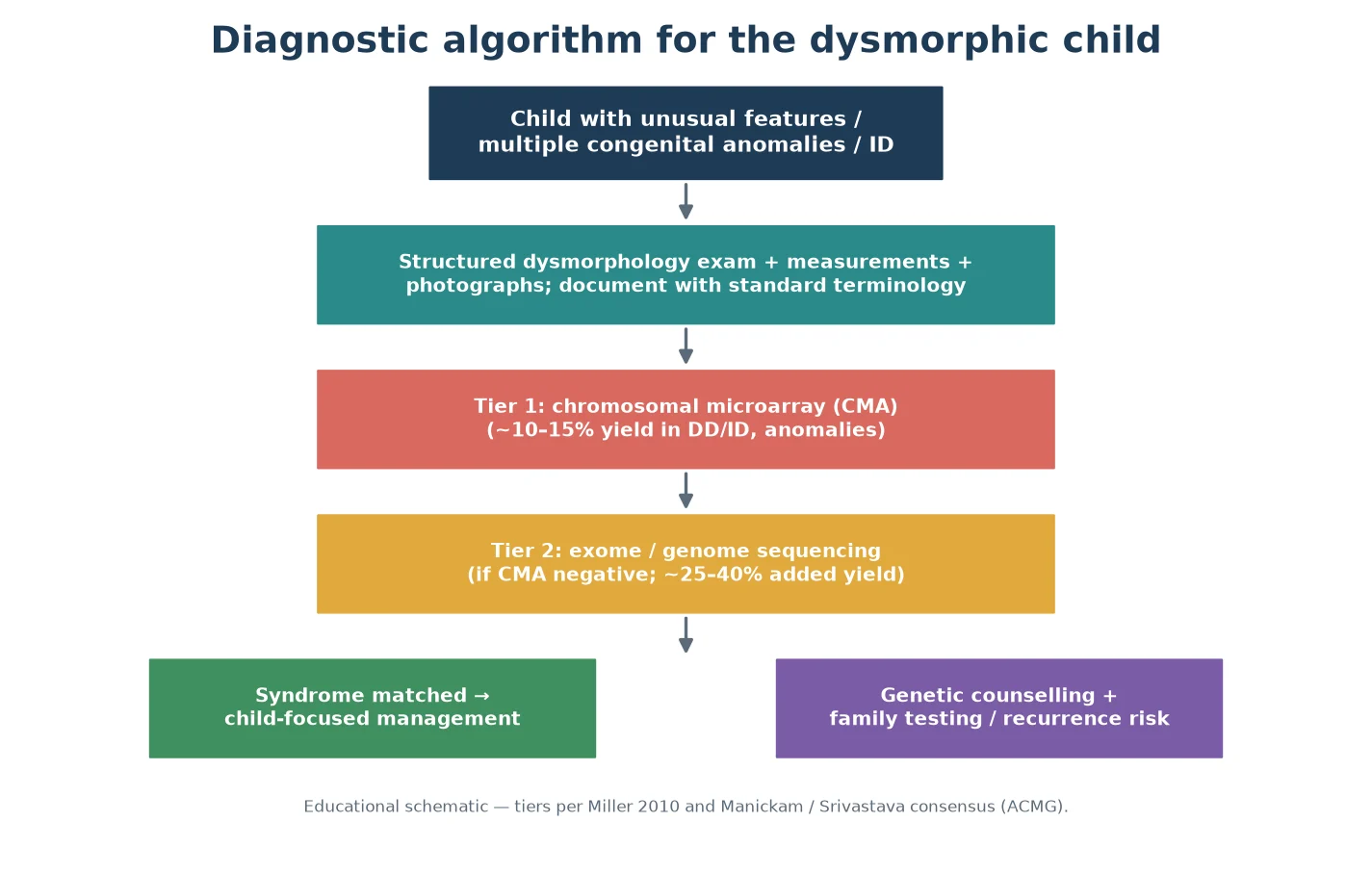
Genetic testing is ordered in a tier, not as a single shotgun. The first-tier test for a child with multiple congenital anomalies, or with developmental delay and intellectual disability, is chromosomal microarray (CMA), because it detects the submicroscopic copy-number changes that a karyotype misses, with a diagnostic yield of roughly ten to fifteen percent in this population. A standard karyotype is now reserved for specific indications such as a suspected balanced rearrangement. [9]
When the microarray is negative and a monogenic syndrome remains likely, escalate to exome or genome sequencing, which adds a substantial increment of diagnosis — on the order of a quarter to two-fifths of previously undiagnosed children in published series. Evidence-based guidelines now position exome and genome sequencing as first-tier for selected children with congenital anomalies or intellectual disability, and the decision to go straight to exome is increasingly defensible at fellowship level. [10] [11]
When a specific syndrome is strongly suspected on clinical grounds, a targeted test is faster and cheaper than a broad search: fluorescence in-situ hybridisation or a quantitative PCR for a 22q11.2 deletion, methylation testing for Prader–Willi or Angelman syndrome, or fragile-X testing where the phenotype or family history fits. Choose the targeted test only when the prior probability is high enough to justify it. [7] [9]
Defining the phenotype fully also guides management, not just diagnosis. Arrange the organ-targeted work-up that the pattern demands — an echocardiogram for a child with a likely chromosomal syndrome, a renal ultrasound for a caudal field defect, hearing and vision assessment for any child with developmental delay, and metabolic testing when the pattern or the decompensation suggests an inborn error. The syndrome label and the organ work-up proceed in parallel. [7] [8]
Management — Resuscitation
In a dysmorphic newborn the immediate priority is airway, breathing and circulation, not the diagnosis. Some syndromic presentations carry time-critical problems that override the work-up: airway obstruction from micrognathia and glossoptosis in Robin sequence may need prone positioning, a nasopharyngeal airway or intubation; a duct-dependent cardiac lesion needs prostaglandin and urgent cardiology; choanal atresia or oesophageal atresia obstruct breathing or feeding and need surgical pathways. Recognise these, stabilise the child, and only then pursue the syndrome. [3] [7]
Management — Definitive & Stepwise
- Perform and document the structured dysmorphology examination with measurements and photography. [1]
- Classify each anomaly by mechanism and significance and generate a syndrome differential. [3] [5]
- Order tier-one chromosomal microarray; add the organ-targeted work-up the pattern demands. [9]
- If the microarray is negative and a monogenic cause is likely, escalate to exome or genome sequencing. [10] [11]
- Confirm a suspected specific syndrome with the targeted test when the prior probability is high. [7]
- Refer to clinical genetics, counsel the family on the result and the recurrence risk, and offer cascade testing of relatives. [8]
- Build the multidisciplinary surveillance plan by organ system, and a clear safety-net and review for the child in whom no diagnosis is yet reached. [7] [8]
Specific Subtypes & Scenarios
Dysmorphic newborn in the delivery suite. Stabilise first, then examine head to toe, measure, photograph, and examine the parents. Send the first-tier microarray and the organ-targeted imaging the pattern demands, and involve clinical genetics early, because a timely diagnosis changes both surgical planning and family counselling. [3] [9]
Older infant or child with delay and an unusual face. This is the classic referral. Combine the dysmorphology examination with a full developmental assessment, request hearing and vision testing, and order tiered genetic testing; the combination of phenotype and developmental trajectory often points to the answer. [7] [8]
Recognised facial gestalt. When the face suggests a known syndrome such as Down, Noonan or Williams syndrome, the examination is used to confirm and to map the complications that follow — the cardiac lesion, the endocrine risk, the developmental profile — so that surveillance begins immediately rather than waiting for the genetic report. [7]
Prenatally suspected syndrome. The postnatal examination confirms, refines or refutes the prenatal suspicion. Compare the findings with the prenatal imaging, confirm measurements against postnatal norms, and communicate clearly with the family, who often arrive with pre-formed fears that need careful framing. [3] [7]
Complications & Pitfalls
The most dangerous pitfall is diagnostic overshadowing: once a syndrome is named, every future symptom gets attributed to it, and treatable disease — an intercurrent infection, a new endocrine problem, pain — is missed. Guard against it by assessing each new presentation on its own merits while holding the syndrome in mind. [7] [8]
Other common errors are avoidable with discipline. Labelling a normal familial or ethnic feature as a syndrome, because no one measured or examined the parents; describing findings in vague non-standard language that no one can interpret later; reassuring a family from a single minor anomaly without searching for a pattern; and rushing to a label before stabilising a sick neonate. Each of these is a recognised viva failure. [1] [5]
An uncertain diagnosis carries its own harm. A family told nothing, or told a frightening guess, may lose trust and disengage from follow-up. The antidote is honest, structured communication: name what you have found, state what testing is underway, give a realistic timeframe, and arrange a definite review point rather than an open-ended wait. [7] [8]
Prognosis & Disposition
Prognosis is driven by the specific syndrome and its complications once identified, and by the organ involvement before a label is reached. Early diagnosis improves outcomes indirectly: it unlocks guideline-based surveillance, secures access to therapy and funding, enables informed family planning, and connects the family to peer support. Even when no diagnosis is made, an explicit plan for periodic review as testing evolves protects the child. [7] [8]
Disposition is shared care. Clinical genetics owns the diagnostic process and counselling; the general paediatric medical home coordinates the whole child; and organ specialists deliver syndrome-specific surveillance. The undiagnosed child is reviewed at set intervals, because re-analysis of stored exome or genome data, and emerging tests, turn previously negative results into answers over time. [10] [11]
Special Populations
Adapt the examination to the child in front of you. The preterm infant and the critically ill neonate may need a staged examination, with the most urgent elements first and the full assessment once stable. In a child from a consanguineous family, weigh autosomal recessive conditions more heavily. In a child from any ethnic background, judge minor variants against the correct population norms, because what is common in one population may be falsely alarming in another. [7] [13]
Communication must be culturally safe. Use professional interpreters when language differs, address stigma head-on by explaining that dysmorphic means an identifiable feature rather than an abnormal child, and frame the plan around the child's strengths and the family's goals. The adolescent with a syndromic diagnosis needs a planned transition to adult care, with genetic and reproductive counselling delivered in an age-appropriate way. [7] [8]
Evidence, Guidelines & Regional Differences
The evidence base for tiered testing is strong and consistent. A 2010 consensus statement established chromosomal microarray as the first-tier test for individuals with developmental disabilities or congenital anomalies, on the strength of its diagnostic yield over karyotyping. [9] A 2021 American College of Medical Genetics and Genomics guideline, and a 2019 meta-analytic consensus, extended the tier upward, positioning exome and genome sequencing as first-tier for selected children with congenital anomalies or intellectual disability. [10] [11]
On the descriptive side, the Elements of morphology project gave the field a shared vocabulary for the head and face, the trunk and limbs, and for general congenital anomaly terms, replacing idiosyncratic descriptions with reproducible ones. [1] [2] [3] [4] Objective facial analysis, from three-dimensional imaging through to deep-learning facial-recognition systems, has shown that machines can discriminate syndromic faces, and these tools now support — but do not replace — the clinical examiner. [6] [12]
Access to clinical genetics, microarray and exome is state- and district-funded but uneven, with longer waits in rural and remote areas and for Māori and Pacific families unless pathways are actively supported. Use local state-funded testing pathways, refer to the nearest clinical genetics service early, and provide cultural-safety and interpreter support so that distance and language do not delay diagnosis. [7]
Genomic medicine is delivered through the NHS Genomic Medicine Service, with documented testing indication codes and a national network of clinical geneticists; NICE and RCPCH guidance shapes referral thresholds. Map the local genomic laboratory centre and its test request process rather than assuming a universal list. [5]
The American College of Medical Genetics and Genomics guidelines drive test selection, and insurance coverage frequently determines which tier is reachable and when; Canadian provincial programmes vary. State the locally funded test and the referral threshold clearly. [9] [10]
Exam Pearls
- Standard terminology comes from the Elements of morphology project — examiners reward correct terms over vague phrases. [1]
- The four mechanism categories — malformation, disruption, deformation, dysplasia — are a perennial viva opener; have an example of each ready. [3]
- Three or more minor anomalies means look hard for a syndrome. [5]
- Chromosomal microarray is first-tier for unexplained developmental delay or multiple congenital anomalies; exome or genome is the next step. [9] [10]
- Robin sequence can be isolated or syndromic — Stickler syndrome is the commonest syndromic cause, so always look. [3]
- Examine the parents; a subtle shared feature changes both diagnosis and recurrence risk. [7]
- Avoid the unexplained word dysmorphic when speaking to families; prefer unique features, then a clear, honest plan. [7]
References
- [1]Allanson JE, Biesecker LG, Carey JC, Hennekam RC Elements of morphology: introduction. American Journal of Medical Genetics Part A, 2009.PMID 19127575
- [2]Allanson JE, Cunniff C, Hoyme HE, McGaughran J, Muenke M, Neri G Elements of morphology: standard terminology for the head and face. American Journal of Medical Genetics Part A, 2009.PMID 19125436
- [3]Hennekam RC, Biesecker LG, Allanson JE, Hall JG, Opitz JM, Temple IK Elements of morphology: general terms for congenital anomalies. American Journal of Medical Genetics Part A, 2013.PMID 24124000
- [4]Biesecker LG, Adam MP, Chung BH, Kosaki K, Menke LA, White SM Elements of morphology: Standard terminology for the trunk and limbs. American Journal of Medical Genetics Part A, 2022.PMID 36062894
- [5]Carey JC, Allanson JE, Hennekam RC, Biesecker LG Standard terminology for phenotypic variations: the elements of morphology project, its current progress, and future directions. Human Mutation, 2012.PMID 22331827
- [6]Hammond P, Hutton TJ, Allanson JE, Buxton B, Campbell LE, Clayton-Smith J Discriminating power of localized three-dimensional facial morphology. American Journal of Human Genetics, 2005.PMID 16380911
- [7]Moeschler JB, Shevell M, American Academy of Pediatrics Committee on Genetics Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics, 2006.PMID 16740881
- [8]Moeschler JB, Shevell M, Committee on Genetics Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics, 2014.PMID 25157020
- [9]Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics, 2010.PMID 20466091
- [10]Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine, 2021.PMID 34211152
- [11]Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genetics in Medicine, 2019.PMID 31182824
- [12]Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D Identifying facial phenotypes of genetic disorders using deep learning. Nature Medicine, 2019.PMID 30617323
- [13]Hoyme HE, May PA, Kalberg WO, Kodituwakku P, Gossage JP, Trujillo PM A practical clinical approach to diagnosis of fetal alcohol spectrum disorders: clarification of the 1996 Institute of Medicine criteria. Pediatrics, 2005.PMID 15629980