Paeds · genetics-dysmorphology-and-metabolism
Genetic history, pedigree construction and inheritance patterns
Also known as Family history taking · Pedigree construction · Inheritance patterns · Mode of inheritance · Recurrence risk · Standardised pedigree nomenclature
A fellowship approach to the genetic family history: draw a standardised three-generation pedigree using National Society of Genetic Counselors nomenclature, recognise each inheritance pattern from its shape (autosomal dominant, autosomal recessive, X-linked, mitochondrial, imprinting, anticipation, multifactorial), apply the recurrence-risk arithmetic that flows from it, and counsel the family honestly and non-directively — including the confounders (variable expressivity, reduced penetrance, gonadal mosaicism, consanguinity, anticipation) that change the numbers.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
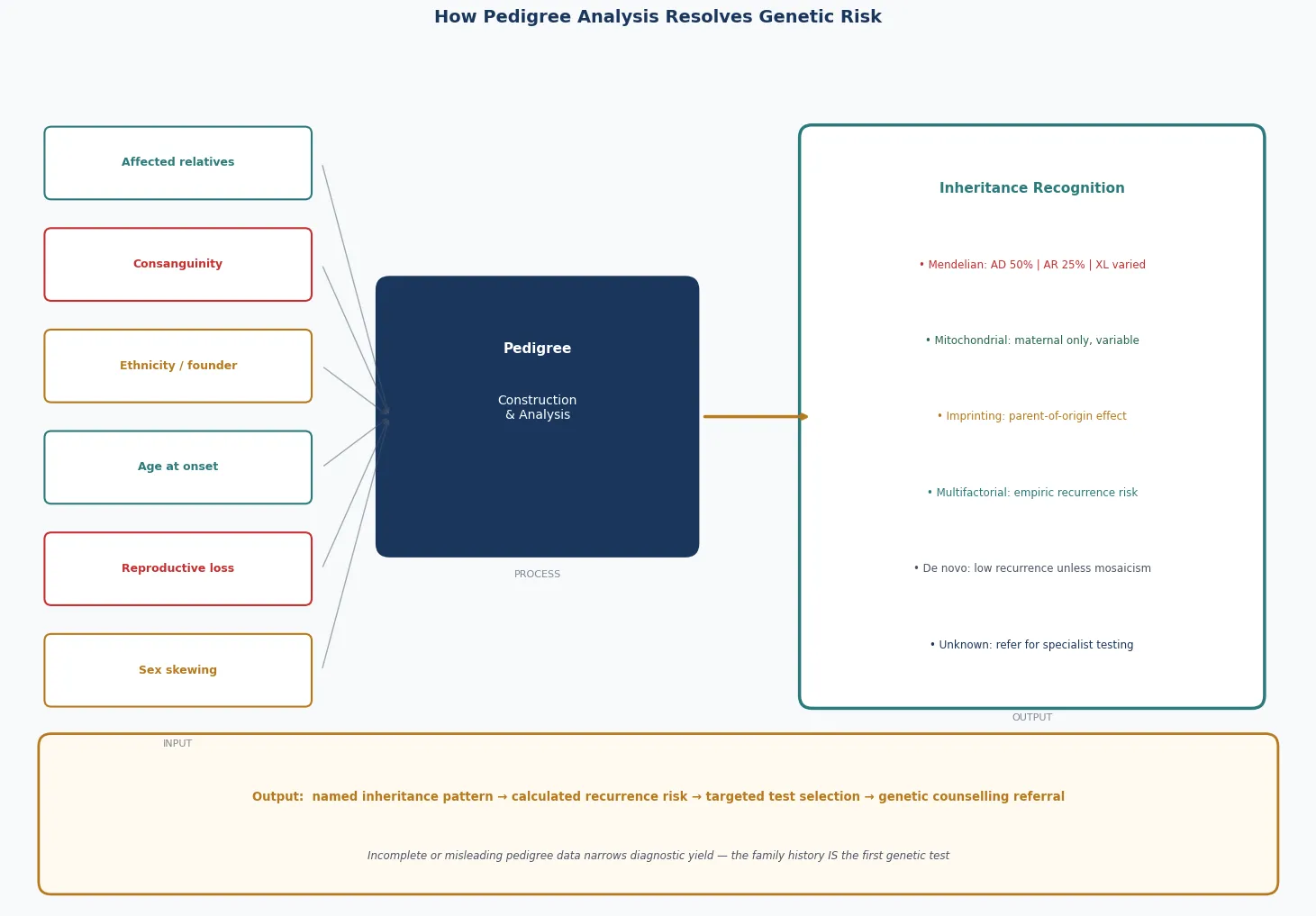
A registrar is asked to assess a three-year-old with global developmental delay and is told, almost as an afterthought, that "there is something in the family." The fellowship task is not to reach for an exome first — it is to sit down and draw the pedigree. Three generations, both parental lines, the consanguinity double-line if it applies, the uncles who died in infancy, the maternal male cousin in a wheelchair, the ethnicity, the miscarriages. From that single sheet of paper the inheritance pattern declares itself, the investigation narrows, and the recurrence risk becomes a number the family can use. The pedigree is the foundation of all of clinical genetics, and it is the skill most often skipped. [1] [2]
4 \u00b7 D.R.A.W. \u2014 the pedigree habit
Overview & Definition
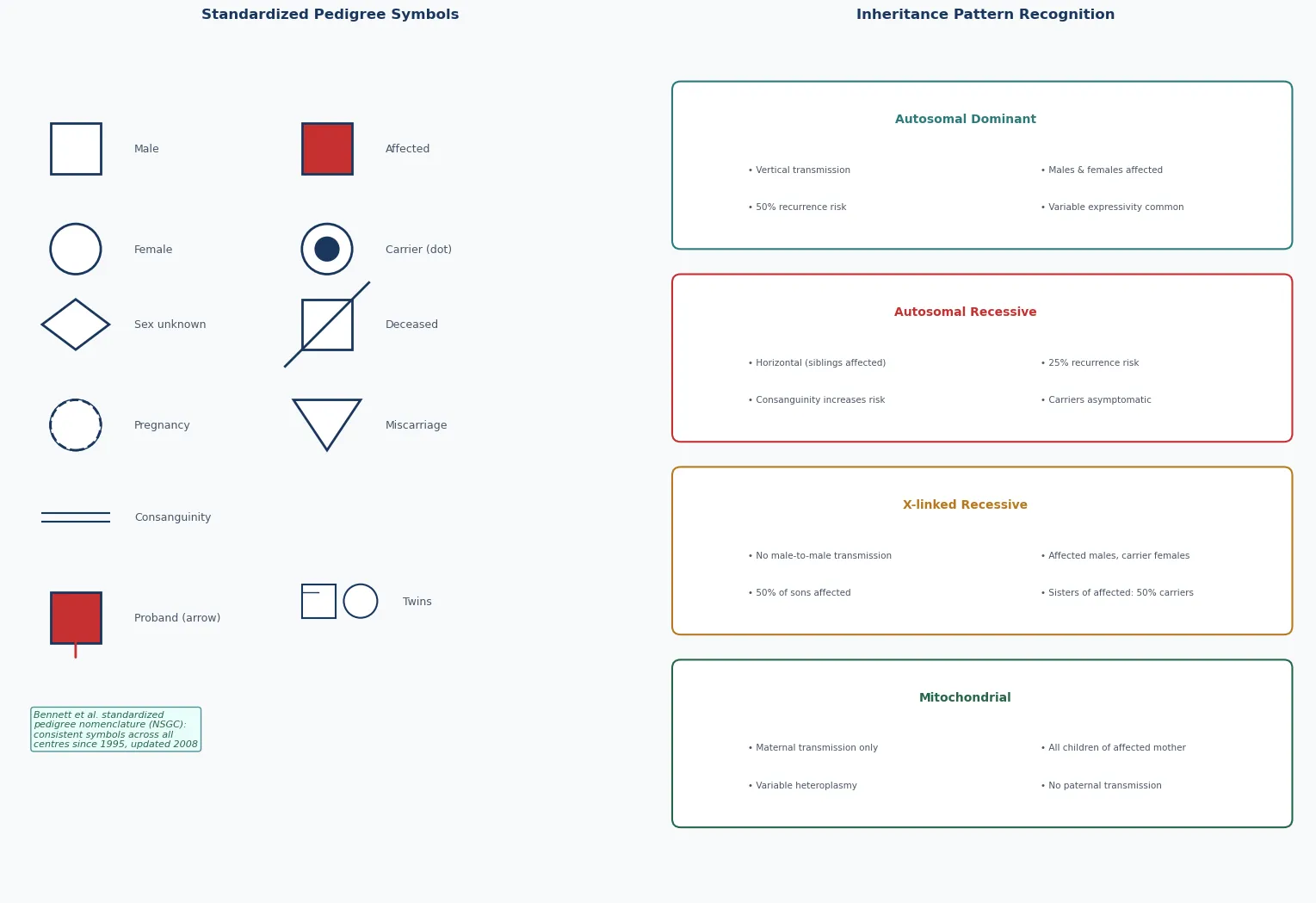
A genetic family history is the structured collection of information about disease in a child's relatives, and a pedigree is its graphical representation, drawn in standardised symbols across at least three generations. The pedigree is not a decoration for the notes — it is a clinical investigation that reorganises a vague sense that "something runs in the family" into a recognisable inheritance pattern, a recurrence risk, and a rational plan for testing and counselling. The National Society of Genetic Counselors standardised pedigree nomenclature precisely so that any clinician, anywhere, could read the same family story from the same symbols. [1] [2]
The reason the pedigree is the foundation of clinical genetics is that the inheritance pattern governs everything that follows — which test to order, what the result means, what the risk is to siblings and future children, and what reproductive options exist. An autosomal dominant family is offered a different conversation from an autosomal recessive family, and both differ from an X-linked family in which the carrier sisters face their own reproductive decisions. Recognising the pattern early prevents the common error of ordering the wrong test, or the right test on the wrong relative. [1] [6]
Inheritance patterns are conventionally grouped into Mendelian (autosomal dominant, autosomal recessive, X-linked dominant, X-linked recessive), non-Mendelian (mitochondrial or maternal, genomic imprinting and parent-of-origin effects, anticipation from trinucleotide-repeat expansion, mosaicism), and multifactorial or polygenic inheritance, with chromosomal disorders forming a parallel category. The same paediatric presentation — developmental delay, a birth defect, a metabolic decompensation — can arise from any of these, and the pedigree is the instrument that distinguishes them. [2] [7]
Classification
Inheritance patterns are classified by where the causal gene lies and how it is transmitted. The Mendelian patterns involve single genes on the autosomes or the X chromosome and follow the segregation ratios that Mendel described. Autosomal dominant disorders are expressed when one copy of a mutant allele is present; autosomal recessive disorders require two. X-linked recessive disorders are expressed in hemizygous males and carried by heterozygous females; X-linked dominant disorders are expressed in both but are often lethal in hemizygous males. These four patterns cover the majority of single-gene paediatric disease. [2] [6]
The non-Mendelian patterns break the Mendelian rules in instructive ways. Mitochondrial inheritance is strictly maternal, because mitochondria are inherited from the oocyte, and it produces the unpredictable variability of heteroplasmy. Genomic imprinting produces disease according to the parent of origin, because methylation silences one allele — so the same 15q11-q13 deletion causes Prader-Willi syndrome when inherited from the father and Angelman syndrome when inherited from the mother. Anticipation describes the worsening severity and earlier onset of trinucleotide-repeat disorders across generations, as the repeat expands. Mosaicism, somatic or gonadal, produces a phenotype that does not obey a simple ratio. [7] [8]
Multifactorial or polygenic inheritance describes the common congenital malformations and adult-onset conditions — neural tube defects, congenital heart disease, cleft lip and palate, diabetes — in which many genes and environment interact, and the recurrence risk is empirical rather than a clean Mendelian ratio. Chromosomal disorders (aneuploidy, translocations, deletions and duplications) form a parallel category: they do not "run in families" in the Mendelian sense, but a balanced translocation in a parent can produce recurrent miscarriage and unbalanced offspring, which is itself a pedigree pattern. Holding the full classification in view is what lets the candidate avoid forcing every family into a single-gene mould. [2] [5]
Epidemiology & Risk Factors
Genetic and congenital disease is common in paediatrics. Roughly two to three per cent of newborns have a recognisable congenital malformation, and mendelian disease accounts for a substantial fraction of chronic paediatric morbidity. When developmental delay, intellectual disability, congenital anomaly, metabolic disease, and familial cancer syndromes are taken together, the genetic family history is relevant in a large minority of every general paediatric clinic. The pedigree is therefore a population-level tool, not a rare-disease curiosity. [2] [4]
The two strongest modifiable risk factors for genetic disease are advancing parental age and consanguinity. Advancing maternal age increases the risk of chromosomal nondisjunction, because oocytes arrest in meiosis I before birth and the meiotic spindle deteriorates over decades — the mechanism behind the age-related rise in trisomy 21 and other aneuploidies. Advancing paternal age increases the risk of new autosomal dominant point mutations, because spermatogonial stem cells divide continuously through life and each division is an opportunity for a copying error — the mechanism behind the paternal-age effect in achondroplasia, Apert syndrome, and other dominant conditions. [3]
Consanguinity is the union of two individuals related by blood, and its genetic significance is that both partners share more alleles than the population average, so their children are more likely to inherit two copies of the same rare recessive allele. Globally, an estimated one in four to one in three marriages in some regions are consanguineous, predominantly first-cousin unions. The offspring of a first-cousin couple carry an additional risk of around two to three per cent of a serious autosomal recessive or multifactorial disorder on top of the population baseline — roughly doubling the background risk — and consanguinity is the single most common reason a previously hidden recessive disorder declares itself in a family. [3] [4]
Population carrier frequency, expressed through the Hardy-Weinberg equilibrium, sets the baseline risk for autosomal recessive disorders in an outbred population. A carrier frequency of one in twenty-five, as for cystic fibrosis in Northern European populations, gives a disease incidence of roughly one in two thousand five hundred live births, because the risk requires both parents to be carriers (one in twenty-five squared) and then a one in four chance per pregnancy. Ethnicity changes the carrier frequency and therefore the screening priority: Tay-Sachs and Gaucher disease in Ashkenazi Jewish families, sickle cell disease in African and Mediterranean populations, beta-thalassaemia in Mediterranean and South Asian populations, and so on. Asking about ethnicity is asking about prior probability. [4] [5]
Pathophysiology
The pathophysiology of each inheritance pattern is rooted in meiosis — the two specialised divisions that halve the chromosome number in gametes and shuffle parental alleles through recombination. Mendelian segregation — the separation of homologous chromosomes in meiosis I — is what produces the clean ratios: an affected heterozygous parent makes equal numbers of mutant and normal gametes, so half the children inherit the allele. Errors in meiosis produce the non-Mendelian patterns: nondisjunction generates aneuploidy, unbalanced recombination generates deletions and duplications, and uniparental disomy (when a child inherits both copies of a chromosome from one parent) can unmask a recessive allele or disrupt imprinting. [2] [7]
Genomic imprinting is the methylation-dependent silencing of one parental copy of a gene, so that only one allele is expressed and the disease phenotype depends on which parent contributed the mutant allele. In the Prader-Willi and Angelman syndrome region at 15q11-q13, the paternal genes are normally expressed and the maternal copy of UBE3A is the active one in the brain. A deletion inherited from the father removes the expressed paternal genes and causes Prader-Willi syndrome; the same deletion inherited from the mother removes the expressed maternal UBE3A and causes Angelman syndrome — two different diseases from the same cytogenetic deletion, explained entirely by parent of origin. [7] [8]
Mitochondrial inheritance is governed by the biology of the mitochondrion. Each cell contains hundreds to thousands of mitochondrial DNA molecules, and a pathogenic variant may be present in some but not all of them — a state called heteroplasmy. Disease appears only when the proportion of mutant mitochondrial DNA exceeds a tissue-specific threshold, and because the proportion is redistributed unpredictably at cell division (mitotic segregation), the severity and the organ involvement vary markedly even within a family. All children of an affected mother may inherit some mutant mitochondria; no children of an affected father do, because mitochondria are transmitted in the oocyte, not the sperm. [2]
Anticipation is the molecular consequence of an unstable trinucleotide-repeat expansion that grows larger across generations. Below a threshold length the repeat is stable; above it, it expands during gametogenesis, and the longer repeat produces earlier onset and more severe disease in the next generation. Fragile X syndrome (a CGG expansion in FMR1), Huntington disease (CAG in HTT), and myotonic dystrophy (CTG in DMPK) are the classic paediatric and young-adult examples, and a pedigree showing worsening severity or earlier onset across generations is anticipation until tested. [2] [7]
Clinical Presentation
Each inheritance pattern presents on the pedigree with a recognisable shape, and a fellowship candidate earns depth by reading the shape rather than memorising a list of diseases. An autosomal dominant pattern shows vertical transmission — affected individuals in every generation, males and females in equal proportion, and the diagnostic feature of male-to-male transmission, which excludes X-linked inheritance. Reduced penetrance and variable expressivity can blur the picture: a mildly affected parent may be overlooked, and a parent who carries the allele but never expresses it can appear to skip a generation. [2] [6]
An autosomal recessive pattern shows horizontal or sibship clustering — multiple affected individuals in a single generation, with unaffected parents who are carriers, and consanguinity over-represented. The onset is often earlier and the phenotype more severe than the dominant counterpart, because a complete loss of function is more damaging than a partial one. The classic counselling for two known carriers is a one-in-four chance per pregnancy, with a two-in-three chance that an unaffected sibling is a carrier. The trap is the isolated case, which is easily mistaken for a de novo change when in fact it is the first presentation of a recessive disorder in a small family. [2] [4]
An X-linked recessive pattern shows affected males connected through carrier females — a characteristic "knight's move" of affected maternal uncles and male cousins, with no male-to-male transmission because a father passes his Y chromosome, not his X, to his sons. Each son of a carrier mother has a one-in-two chance of being affected, and each daughter has a one-in-two chance of being a carrier; the overall risk to a child of a carrier mother is one in four. An X-linked dominant pattern differs in that heterozygous females are affected and hemizygous males are often so severely affected that they do not survive — producing a pedigree with affected females, fewer affected males, and an excess of miscarriages. [2] [10]
A mitochondrial pattern shows maternal-only transmission — all the children of an affected mother may be affected to varying degrees, and none of the children of an affected father are. The variability is the hallmark: within a single sibship, one child may have a severe encephalomyopathy while another has only a mild ptosis, because the proportion of mutant mitochondrial DNA differs between tissues. An imprinting pattern shows parent-of-origin dependence — the same cytogenetic change produces one disease from the father and another from the mother — and an anticipation pattern shows worsening severity or earlier onset across generations. [7] [2]
Differential Diagnosis
The differential diagnosis of an inheritance pattern is the set of confounders that make one pattern look like another, and the discriminating skill is to know which confounder is operating. The four most common are reduced penetrance, variable expressivity, anticipation, and gonadal mosaicism — and each has a signature that the candidate should be able to name. [6] [9]
Reduced penetrance means that not everyone who carries a dominant allele expresses the disease, so the pedigree appears to skip a generation and can be misread as recessive or as a phenocopy. Variable expressivity means that carriers express the disease to different degrees, so a mildly affected parent is overlooked and a parent and child appear to have different conditions. The contemporary reappraisal of these concepts emphasises that they are not exceptions but the rule in the genomic era, because most pathogenic variants produce a spectrum rather than a binary phenotype, and a counselling conversation that ignores them will misquote the risk. [6]
Anticipation makes a dominant disorder look as though it is worsening across generations, because the trinucleotide repeat expands — a grandparent with a late-onset, mild phenotype, a parent with an earlier onset, and a child with a severe childhood disease, all from the same gene. Gonadal (germline) mosaicism explains the recurrence of a de novo-dominant condition in a family in which neither parent is somatically affected: one parent carries the mutation in a proportion of their gametes, and the recurrence risk is higher than the population baseline. Non-paternity, phenocopy (an environmentally caused mimic of a genetic disease), and early death that removes affected individuals from view all distort the pedigree further. [9] [10]
| Feature | Autosomal dominant | Autosomal recessive (small family) |
|---|---|---|
| Parent and child affected | Expected (vertical) | Unlikely unless assortative mating or a parent homozygous/compound |
| Consanguinity | Not predictive | Strongly predictive |
| Sibship recurrence | Each sibling 1 in 2 | Each sibling 1 in 4 |
| Confounder to consider | Reduced penetrance, variable expressivity | De novo, parental mosaicism, phenocopy |
Clinical & Bedside Assessment
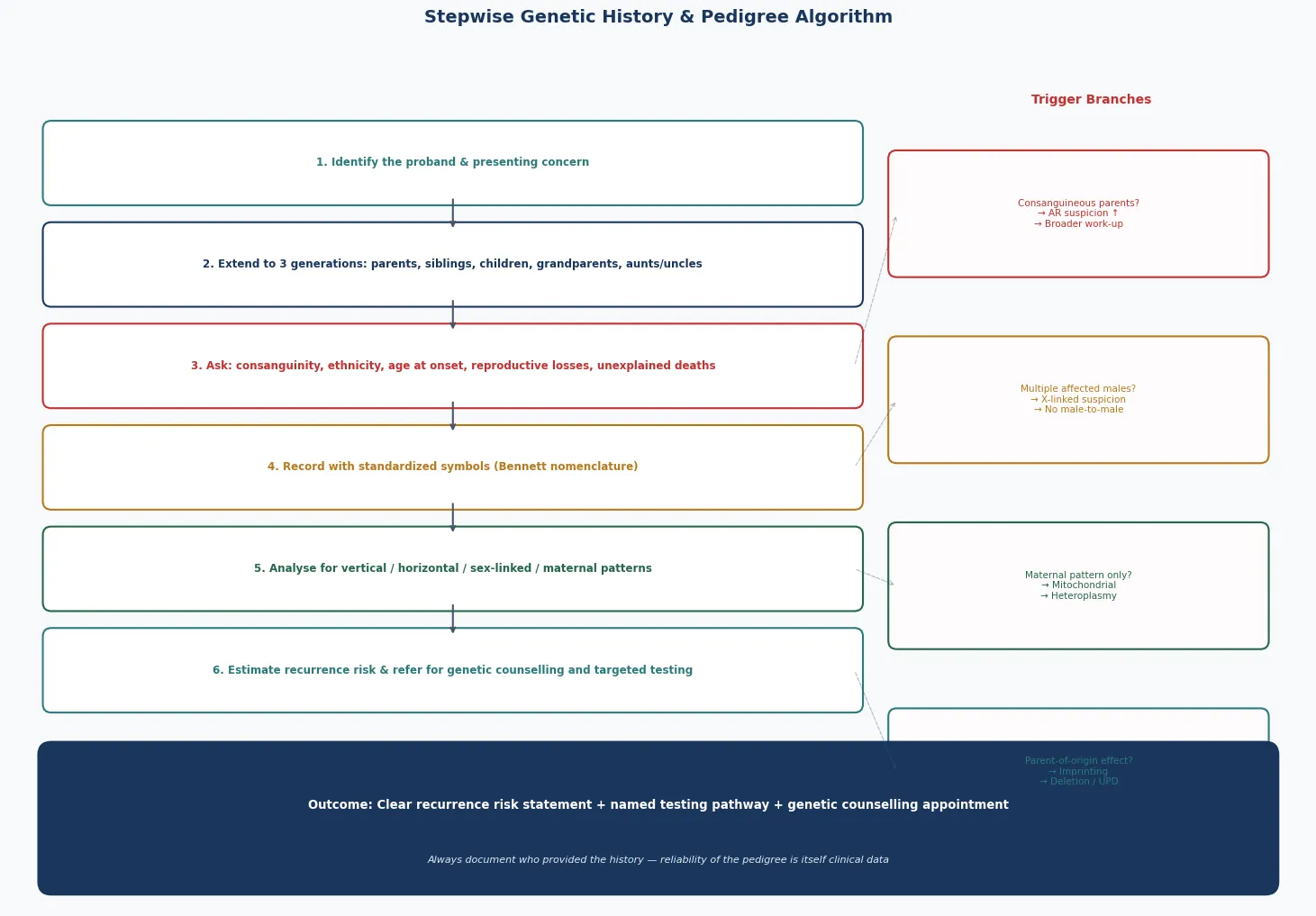
The bedside assessment begins with the history and is completed by the pedigree, and the order matters. Ask first about the presenting child, then widen to the siblings, then to both parents and their siblings, then to the grandparents, aiming for at least three generations on both the maternal and the paternal side. Record the ethnicity of each line, ask directly and respectfully about consanguinity (whether the parents are related by blood, and if so how), and document every pregnancy outcome including miscarriages, stillbirths, and infant deaths — because recurrent loss is itself a pedigree pattern, often signalling a balanced translocation or a recessive lethal. [1] [2]
Draw the pedigree in standardised National Society of Genetic Counselors symbols so that any geneticist can read it. A square is a male, a circle is a female, a diamond is of unknown or unspecified sex; a fully shaded symbol is affected, a half-shaded symbol or a central dot denotes a carrier where carrier status is defined, a diagonal line through a symbol denotes deceased, and an arrow identifies the proband. A horizontal line joins a couple, a vertical line drops to their children, a double horizontal line denotes consanguinity, and identical and fraternal twins have their own symbols. Pregnancy, pregnancy loss, and termination each carry a defined symbol. The discipline of drawing, rather than narrating, forces the clinician to confront gaps and contradictions in the history. [1]
A set of family-history red flags should prompt formal genetics referral and a more searching pedigree. Two or more close relatives with the same condition, a condition appearing at an unusually early age, a known or suspected consanguineous union, an ethnic predilection for a specific disease, recurrent miscarriages or stillbirths, unexplained intellectual disability or developmental regression, a sudden unexplained death in the young, and a recognised familial cancer syndrome are all red flags. The paediatrician's job is to recognise the flag and draw the pedigree, not to deliver the final diagnosis — that is a shared task with clinical genetics. [5] [4]
Use the pedigree at the bedside to choose the investigation rather than guessing. A chromosomal suspicion (multiple congenital anomalies, a dysmorphic syndrome, recurrent miscarriage) points to a karyotype or chromosomal microarray. An unexplained developmental delay points to a microarray first and then an exome or genome if the microarray is normal. A recognised single-gene pattern points to a targeted test or gene panel, and an imprinting suspicion (Prader-Willi, Angelman) points to methylation-specific testing rather than a generic microarray. The right test for the right pattern is the single biggest determinant of a useful result. [7] [1]
Investigations
The investigation plan in clinical genetics is governed by the inheritance pattern recognised on the pedigree. The first principle is to test the affected individual first — an affected proband is far more informative than a predictive test on an unaffected relative, because a negative result in an unaffected relative is uninformative if the family variant is unknown. The second principle is to match the test to the suspected mechanism: a karyotype for a balanced translocation, a chromosomal microarray for an unbalanced copy-number change, methylation studies for an imprinting disorder, and a single-gene test, gene panel, or exome/genome for a single-gene disorder. [2] [7]
Methylation studies are the key investigation for imprinting disorders such as Prader-Willi and Angelman syndromes, because they detect the parent-of-origin methylation abnormality directly, whether it arises from a deletion, uniparental disomy, or an imprinting-centre mutation. A chromosomal microarray will detect the deletion but cannot, by itself, establish the parent of origin, which is why methylation testing is the preferred first-line investigation for these syndromes. Once a pathogenic variant is identified in a family, cascade testing of at-risk relatives — carrier testing, predictive testing, and prenatal testing — is organised through clinical genetics. [7]
Biochemical testing complements DNA testing, particularly for inherited metabolic disease. Enzyme assays and metabolite measurement confirm many inborn errors of metabolism, the newborn bloodspot screen detects a defined set of treatable disorders in the first days of life, and both guide the interpretation of variants of uncertain significance identified on exome or genome sequencing. The pedigree informs which biochemical tests are worth sending: a consanguineous family raises the prior probability of a recessive metabolic disorder and lowers the threshold to send an acylcarnitine profile, a plasma amino acid quantitation, and a urine organic acid analysis. [4] [2]
Management — Resuscitation
Resuscitation in the genetic context means acting on the family-history findings that demand an urgent response rather than elective referral. Three scenarios dominate paediatric practice: an acutely unwell consanguineous neonate with a suspected inborn error of metabolism, a family history of sudden unexplained death, and a known X-linked or other lethal condition in a current pregnancy. Each has a defined pathway, and in each the pedigree informs the immediate management. [4] [5]
A neonate born to consanguineous parents who presents with encephalopathy, metabolic acidosis, hypoglycaemia, hyperammonaemia, or liver dysfunction is an inborn error of metabolism until excluded. The immediate management is to halt catabolism — stop feeds, give intravenous glucose and fluid, and treat presumptively with the relevant emergency measures (for example, nitrogen-scavenging therapy if a urea-cycle disorder is suspected) — while sending the metabolic and genetic work-up. The pedigree sharpens the prior probability toward an autosomal recessive metabolic disorder and should accelerate both the work-up and the involvement of the metabolic team. [4]
A family history of sudden unexplained death, particularly in a young person, raises inherited cardiac disease — the channelopathies (long QT, Brugada, catecholaminergic polymorphic ventricular tachycardia) and the cardiomyopathies (hypertrophic, dilated, arrhythmogenic right ventricular). The immediate response is cardiology assessment of the proband and the at-risk relatives, with an electrocardiogram and echocardiogram, and referral for genetic testing where the family pattern fits. Recognising the inheritance pattern on the pedigree — most are autosomal dominant — directs cascade testing and may prevent a further death. [2]
Taking the pedigree early, before the family disperses after a death or a critical illness, is itself a time-critical step. Relatives are contactable, memories are fresh, and the consanguinity and ethnicity that sharpen the diagnosis are easiest to capture in the acute window. A pedigree begun in the emergency department or the neonatal unit is a far richer document than one reconstructed weeks later, and it can change the immediate differential diagnosis. [5] [4]
Management — Definitive & Stepwise
Definitive management of a suspected genetic condition follows a stepwise pathway that the pedigree frames at every stage: confirm the diagnosis, establish the inheritance pattern, calculate the recurrence risk, offer carrier and predictive testing, and refer for formal genetic counselling. Each step has a defined purpose, and skipping a step — most often the formal pedigree and counselling — is the commonest source of error. [1] [2]
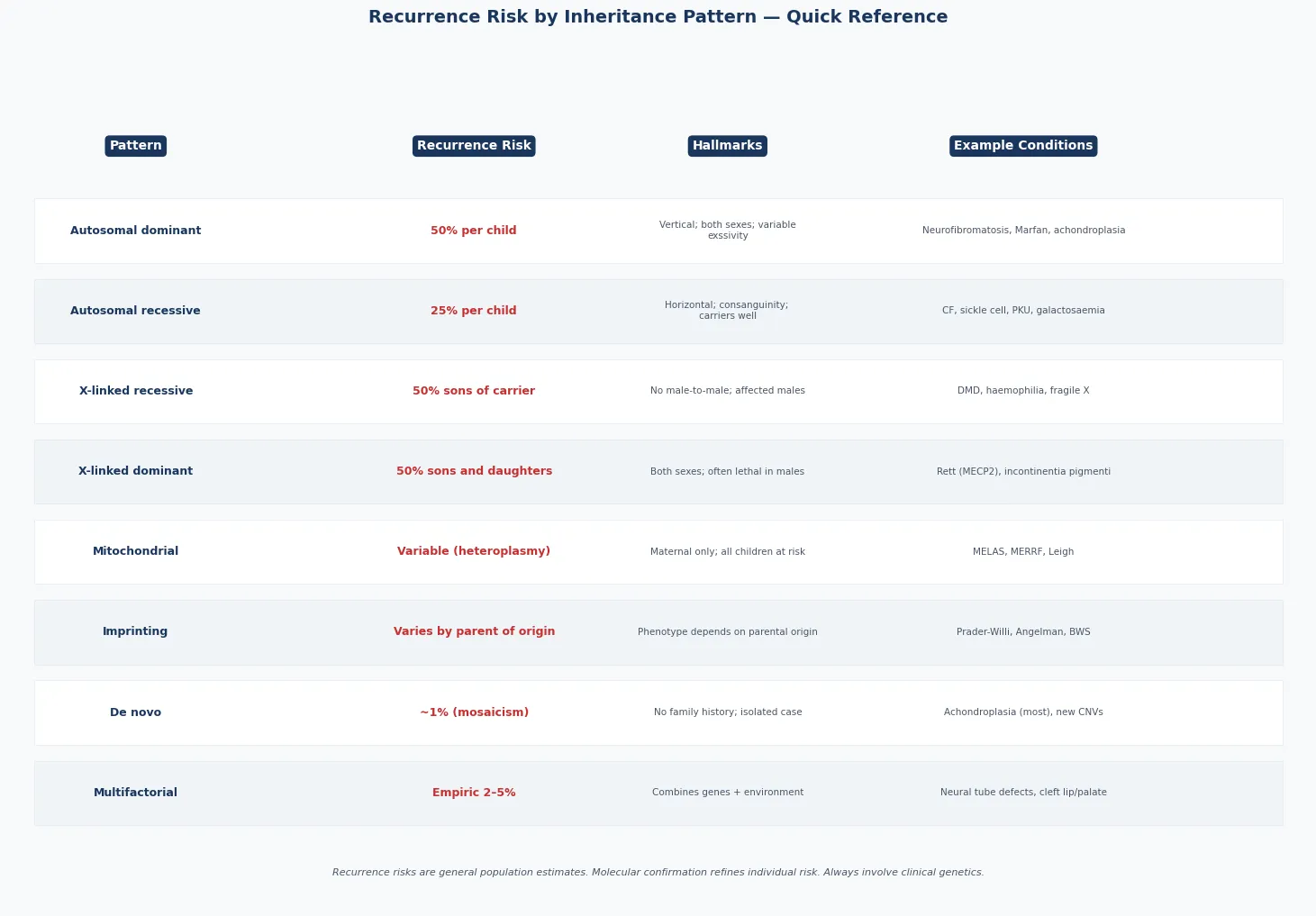
The recurrence risk is communicated in the language the family can use. For autosomal dominant, the risk to a child of an affected heterozygous parent is one in two. For autosomal recessive, the risk to a child of two carriers is one in four. For X-linked recessive, the overall risk to a child of a carrier mother is one in four (one in two for each son). These Mendelian ratios are modified by the confounders: reduced penetrance lowers the risk of overt disease, variable expressivity broadens the phenotype, gonadal mosaicism raises the recurrence risk of a "de novo" change above the population baseline, and consanguinity raises the prior probability of recessive disease. Bayesian adjustment incorporates prior information — for example, a woman with an affected brother and a normal creatine kinase may still be a Duchenne carrier, and her risk is calculated by combining the pedigree prior with the test result. [2] [10]
The reproductive options that follow from a recognised recurrence risk are prenatal diagnosis (chorionic villus sampling or amniocentesis), preimplantation genetic testing with in-vitro fertilisation, donor gametes, adoption, and the decision to take the risk or to have no further children. The clinician's role is to lay out the options honestly and non-directively, not to choose for the family, and to recognise that the right choice depends on the family's values, their experience of the disease, and their access to the technology. Formal genetic counselling is the forum for this conversation. [1] [2]
The family is enrolled in cascade and surveillance testing through a named coordinator, who arranges carrier testing of at-risk relatives, predictive testing where appropriate, and the paediatric and adult surveillance that the diagnosis requires. The coordinator is the safeguard against fragmented care, because a genetic diagnosis propagates across a family and across a lifetime, and a single missed relative can mean a missed opportunity for prevention. The general paediatrician remains the medical home, holding the surveillance schedule and coordinating the subspecialty input. [5] [1]
Specific Subtypes & Scenarios
Each inheritance pattern carries a distinctive counselling scenario, and a fellowship answer earns depth by handling them individually rather than as a single block. The patterns generalise, but the counselling is pattern-specific. [1] [2]
The autosomal dominant scenario centres on the family in which the disease is present in every generation. The counselling addresses the one-in-two risk, but also the confounders that make the phenotype unpredictable: variable expressivity means a child's disease may be milder or more severe than the parent's, and reduced penetrance means a carrying parent may never develop the disease at all. Neurofibromatosis type 1, Marfan syndrome, achondroplasia, and tuberous sclerosis are the paediatric examples, and the contemporary reappraisal of penetrance and expressivity emphasises that the one-in-two ratio is a transmission risk, not a severity prediction. [6]
The autosomal recessive scenario centres on the consanguineous family or the family with two unaffected carrier parents and one affected child. The counselling addresses the one-in-four risk, the two-in-three carrier probability for unaffected siblings, and the option of carrier testing of the wider family. Cystic fibrosis, spinal muscular atrophy, phenylketonuria, and congenital adrenal hyperplasia are the paediatric examples, and consanguinity is the single most common reason a recessive disorder declares itself. The consanguinity conversation is framed as a risk modifier, not a blame. [4] [5]
The X-linked recessive scenario centres on the affected male and the carrier females who face their own reproductive decisions. The counselling addresses the one-in-four overall risk to a carrier mother's children, the absence of male-to-male transmission, and the manifesting female — explained by skewed X-inactivation, a structural chromosomal rearrangement, or uniparental disomy. Duchenne muscular dystrophy and haemophilia are the paediatric examples, and the isolated male with a de novo change still carries a recurrence risk above baseline because of gonadal mosaicism in the mother. [10] [9]
The non-Mendelian scenario centres on the patterns that break the Mendelian rules. A mitochondrial disorder is recognised by maternal-only transmission and unpredictable intrafamilial variability, and the counselling addresses the impossibility of predicting severity from the genotype alone. An imprinting disorder such as Prader-Willi or Angelman syndrome is recognised by the parent-of-origin dependence, and the counselling addresses the mechanism — deletion, uniparental disomy, or imprinting-centre mutation — because each carries a different recurrence risk. An anticipation disorder such as fragile X is recognised by worsening severity across generations, and the counselling addresses the premutation-carrier risk in the mother. [7] [2]
Why germline mosaicism is the counselling trap in an isolated case
An isolated child with a confirmed de novo dominant or X-linked change is too readily dismissed as "sporadic, no recurrence risk." In reality, one parent may carry the mutation in a proportion of their gonadal cells — germline (gonadal) mosaicism — and the recurrence risk is therefore higher than the population baseline, often quoted at around one per cent but sometimes substantially more. Large studies of phased de novo variants and registries of Duchenne muscular dystrophy confirm that germline mosaicism accounts for a measurable fraction of recurrence, and the safe counselling is to offer prenatal diagnosis in any subsequent pregnancy regardless of the parents' somatic testing. [9] [10]
Complications & Pitfalls
The complications of poor pedigree practice are the missed diagnoses, the misquoted recurrence risks, and the wrong tests that follow. A fellowship answer handles both the patterns that are missed and the assumptions that cause them, because the harm in clinical genetics is rarely the genetic change itself — it is the counselling error or the missed opportunity for prevention. [1] [2]
The dominant-versus-recessive pitfall arises when a parent and child are both affected and the candidate assumes dominant without considering an autosomal recessive disorder in a small family, a parent who is homozygous or compound heterozygous, or assortative mating in a population with a high carrier frequency. Conversely, a dominant disorder with reduced penetrance can be misread as recessive when a carrying parent never expresses the disease. The pedigree, read with the confounders in mind, prevents both errors. [6]
The X-linked pitfall arises when affected maternal uncles or male cousins are overlooked, because they are not the proband's immediate family, and a pattern that should declare itself is hidden. The converse error is to overlook the manifesting female, who is explained by skewed X-inactivation rather than by a wrong diagnosis, and who may carry a recurrence risk of her own. Asking specifically about the maternal uncles and male cousins is the single most rewarding pedigree habit in suspected X-linked disease. [10]
The consanguinity pitfall arises when consanguinity is never asked about, because the clinician is uncomfortable raising it. The consequence is a missed autosomal recessive diagnosis, a misquoted recurrence risk, and a lost opportunity for carrier testing of the wider family. Consanguinity is a population-level reality across many ANZ communities, and the conversation — framed as a risk modifier, conducted respectfully, and never as a blame — is a core paediatric skill. [4] [5]
The test-selection pitfall arises when the wrong investigation is ordered for the pattern: a single-gene test when a panel or exome is indicated by an unsolved or heterogeneous phenotype, or conversely an exome when a methylation study or karyotype would have answered the question more cheaply and more quickly. Matching the test to the pattern, and to the family's counselling needs, is the safeguard. [7] [2]
Prognosis & Disposition
The prognosis of a genetic condition is set partly by the disease and partly by the inheritance pattern, which determines the surveillance plan for the child and the cascade testing for the wider family. A fellowship answer frames prognosis in both dimensions, because the family needs to know not only what the future holds for the affected child but also what it holds for the siblings, the parents, and the unborn children. [2] [1]
The determinants of prognosis include the natural history of the specific disease, the response to treatment where one exists, and the timeliness of the surveillance that catches the preventable complications. A consanguineous family with a confirmed autosomal recessive disorder benefits from cascade carrier testing, which can inform the reproductive decisions of the unaffected siblings and the wider extended family, and from the early diagnosis of future affected children through prenatal or newborn testing. The prognosis is therefore not fixed by the genotype; it is shaped by the quality of the family-wide surveillance. [4] [5]
Predictive testing in a child is governed by a defined ethical principle: testing is offered in childhood for an actionable childhood-onset disorder, but deferred to the competent adult for an adult-onset condition, because the child's autonomy and their future right not to know are respected. Huntington disease is the classic example of a condition for which predictive testing in a minor is not offered; familial hypercholesterolaemia, in which childhood treatment changes outcome, is the classic example of one for which it is. The counselling conversation, not the blood test, is the heart of this decision. [2] [6]
Disposition for a general paediatrician is shared, structured care. The paediatrician recognises the pattern, draws the pedigree, coordinates the investigation and surveillance, and refers to clinical genetics for formal counselling, while remaining the medical home across the lifespan. The named coordinator arranges cascade testing and the surveillance schedule, and the subspecialty input is invoked at the relevant point. The principles are constant; the delivery adapts to the family. [1] [2]
Special Populations
The genetic family history interacts with the child's social, cultural, and developmental context, and the same inheritance pattern behaves differently across populations. Access, understanding, and cultural framing all shape the counselling, and a fellowship answer recognises that the pedigree is only as good as the family's ability to engage with it. [4] [1]
Consanguineous families and communities with a tradition of endogamy carry an elevated risk of autosomal recessive disease, and the counselling is framed as a risk modifier rather than a prohibition. The conversation is conducted respectfully and without stigma, the increased risk (an additional two to three per cent for a first-cousin union) is quantified honestly, and the option of carrier screening for the disorders prevalent in the community is offered. The aim is to support the family's reproductive autonomy while giving them the information to act on it. [3] [4]
Ethnicity shapes carrier-screening priorities across ANZ. Tay-Sachs and Gaucher disease in Ashkenazi Jewish families, sickle cell disease in African and Mediterranean populations, beta-thalassaemia in Mediterranean and South Asian populations, cystic fibrosis in Northern European populations, and spinal muscular atrophy across many populations are the classic examples. Asking about ethnicity is asking about prior probability, and the carrier screen offered to a couple is informed by their background as well as by their family history. [5] [4]
Migrant, refugee, and asylum-seeking families may present with an unrecognised inherited disorder and no documented family history, having had no antenatal screening and no access to genetics services in their country of origin. A careful reconstruction of the pedigree with interpreter support, confirmation of the diagnosis and the mechanism, and a written explanation in the family's language are the foundations. Consanguinity, ethnicity, and exposure to infectious or environmental triggers are all part of the reconstructed history. [1] [4]
Indigenous and remote-community families may face extended family structures, a higher burden of certain conditions, and limited access to genetics services by distance. Telehealth and outreach extend pedigree construction, counselling, and cascade testing into these settings, and the pedigree is drawn with an awareness that family relationships may be described differently and that the biological and the social family may not coincide. The principles of respectful, non-directive counselling are constant; the delivery adapts to the setting. [2] [5]
Evidence, Guidelines & Regional Differences
The evidence base for pedigree practice rests on the standardised nomenclature of the National Society of Genetic Counselors, on the population genetics of consanguinity, and on the contemporary reappraisal of penetrance and expressivity in the genomic era. The foundational references are operational — they tell the clinician how to draw the pedigree and how to read it — and a fellowship candidate should be able to name them. [1] [2]
The Bennett 1995 paper in the American Journal of Human Genetics established the standardised human pedigree nomenclature through the Pedigree Standardization Task Force, and the Bennett 2008 update in the Journal of Genetic Counseling reviewed and assessed those recommendations in the light of a decade of use. Together they define the symbols, the conventions, and the rules that allow any clinician to read any pedigree, and they are the operational standards around which most clinical genetics services build their practice. [2] [1]
The consanguinity evidence is led by the work of Bittles and colleagues, who quantified the population genetics and the health impact of inbreeding across global communities, and by clinical studies such as Shieh and colleagues on consanguinity and the risk of congenital heart disease, which provide the worked clinical examples of the autosomal recessive risk shift. The contemporary reappraisal of reduced penetrance and variable expressivity is captured in the 2024 American Society of Human Genetics presidential address, which argues that these are the rule rather than the exception in the genomic era and that they must inform every counselling conversation. [3] [6]
Regional differences in ANZ practice are practical rather than scientific. Australia and New Zealand apply the international pedigree and counselling standards with a national newborn bloodspot screening programme, publicly funded clinical genetics services, and increasing access to exome and genome sequencing for undiagnosed paediatric disease. Culturally competent, non-stigmatising counselling for consanguineous couples is a particular priority given the diversity of migrant and endogamous communities, and telehealth and outreach extend the service into rural and remote settings. The principles are constant; the delivery adapts to the setting. [4] [1]
In Australia and New Zealand, the genetic family history is taken and the pedigree is drawn in standardised National Society of Genetic Counselors nomenclature across at least three generations, with direct and respectful inquiry about consanguinity and ethnicity. Publicly funded clinical genetics services provide formal counselling and advanced testing, and the national newborn bloodspot screen detects a defined set of treatable inherited conditions. Telehealth and outreach extend pedigree construction, counselling, and cascade testing into rural and remote communities, and culturally competent counselling for consanguineous couples frames consanguinity as a risk modifier rather than a stigma. [1] [4]
Exam Pearls
A fellowship candidate answering on genetic history, pedigree construction, and inheritance patterns should land five anchor points and avoid four classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [1] [2]
Anchor one: draw the pedigree. A standardised three-generation pedigree, in National Society of Genetic Counselors symbols, is the cheapest and often the most informative genetic test. Draw it before you order a test, and document both parental lines, the consanguinity, the ethnicity, the pregnancy losses, and the causes of death. [1]
Anchor two: recognise the pattern from the shape. Vertical transmission is autosomal dominant, horizontal or sibship clustering with unaffected parents is autosomal recessive, affected males through carrier females with no male-to-male transmission is X-linked recessive, and maternal-only transmission is mitochondrial. The shape is the diagnosis. [2]
Anchor three: apply the recurrence-risk arithmetic. One in two for autosomal dominant, one in four for autosomal recessive, one in four overall for X-linked recessive — then modify for penetrance, mosaicism, consanguinity, and Bayesian prior information. A flat risk quoted without the modifiers is a counselling error. [6] [10]
Anchor four: ask directly about consanguinity and ethnicity. A first-cousin union roughly doubles the baseline risk of a congenital or genetic disorder and reframes the prior probability toward autosomal recessive disease, and ethnicity sets the carrier-screening priority. Asking is a core paediatric skill, conducted respectfully and without stigma. [3] [4]
Anchor five: counsel non-directively and refer. Lay out the reproductive options honestly, support the family's autonomous decision, and refer for formal genetic counselling whenever the pattern is complex or the risk is high. The paediatrician remains the medical home; clinical genetics owns the complex counselling. [1] [2]
The four traps to avoid are assuming dominant from two affected generations without considering reduced penetrance and recessive disease, overlooking the affected maternal uncles that reveal an X-linked pattern, quoting a flat recurrence risk without the confounders, and failing to ask about consanguinity and ethnicity. Avoid these and the rest of the answer falls into place. [1] [6]
References
- [1]Bennett RL, French KS, Resta RG, Doyle DL. Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors. J Genet Couns, 2008.PMID 18792771
- [2]Bennett RL, Steinhaus KA, Uhrich SB, et al. Recommendations for standardized human pedigree nomenclature. Pedigree Standardization Task Force of the National Society of Genetic Counselors. Am J Hum Genet, 1995.PMID 7887430
- [3]Bittles AH, Black ML. Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci U S A, 2010.PMID 19805052
- [4]Bittles AH. Endogamy, consanguinity and community disease profiles. Community Genet, 2005.PMID 15767749
- [5]Shieh JT, Bittles AH, Hudgins L. Consanguinity and the risk of congenital heart disease. Am J Med Genet A, 2012.PMID 22488956
- [6]Gelb BD. 2024 ASHG presidential address: Incomplete penetrance and variable expressivity: Old concepts, new urgency. Am J Hum Genet, 2025.PMID 40054435
- [7]Butler MG. Clinical Presentation, Genetics, and Laboratory Testing with Integrated Genetic Analysis of Molecular Mechanisms in Prader-Willi and Angelman Syndromes: A Review. Int J Mol Sci, 2026.PMID 41683698
- [8]Ryan NM, Heron EA. Evidence for parent-of-origin effects in autism spectrum disorder: a narrative review. J Appl Genet, 2023.PMID 36710277
- [9]Lecoquierre F, Drouot N, Coutant S, et al. Parental germline mosaicism in genome-wide phased de novo variants: Recurrence risk assessment. PLoS Genet, 2025.PMID 40163539
- [10]Verebi C, Gravrand V, Bienvenu T, et al. A retrospective cohort study and review of the literature about germline mosaicism in Duchenne muscular dystrophy. J Genet Couns, 2025.PMID 38895972