Paeds · respiratory-sleep-and-airway
Congenital lung and airway malformations
Also known as Congenital lung malformations · Congenital thoracic malformations · Congenital pulmonary airway malformation (CPAM/CCAM) · Bronchopulmonary sequestration · Congenital lobar emphysema · Bronchogenic cyst
Fellowship guide to congenital lung and airway malformations — the CPAM, sequestration, lobar emphysema, bronchogenic cyst and bronchial atresia spectrum, how antenatal detection has rewritten the epidemiology, the two questions that drive management (is it causing trouble now, and does an asymptomatic lesion need surgery or surveillance?), and the malignancy risk that keeps the resection debate alive.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Related topics
Overview & Definition
A healthy-looking newborn is reviewed on the postnatal ward because the twenty-week scan showed a bright patch in one lung. The baby feeds well, breathes comfortably, and looks entirely normal, yet the parents have spent half the pregnancy fearing the worst. The paediatric task is to explain what the lesion is, decide what imaging and follow-up it needs, and give a straight answer about whether an operation is coming — and this scenario, the well baby with an antenatal finding, is now the commonest way these lesions present. [1] [8]
Congenital lung and airway malformations are abnormalities of lung or large-airway development that arise before birth and range from a tiny incidental cyst to a life-threatening mass that fills the chest. The named lesions overlap in their embryology and imaging, and hybrid forms that combine features of two lesions are common, so it is more useful to treat them as one spectrum with shared principles than as rigidly separate diseases. [1] [2]
What has genuinely changed the topic is antenatal ultrasound. A generation ago most lesions declared themselves through neonatal distress or recurrent childhood infection; today the majority are detected in an asymptomatic fetus and followed to a planned, calm postnatal pathway. This shift explains why so much of the modern literature and so many of the hardest questions are about the child who has no symptoms at all. [8] [1]
Classification
Sorting these lesions starts with a single anatomical question that cuts the spectrum in two: does the abnormal tissue draw its blood from the pulmonary circulation like normal lung, or from a systemic artery arising straight off the aorta? A systemic arterial supply is the fingerprint of bronchopulmonary sequestration, and finding that feeding vessel on imaging both makes the diagnosis and warns the surgeon what to expect. Everything else in the spectrum keeps a normal pulmonary blood supply. [1] [11]
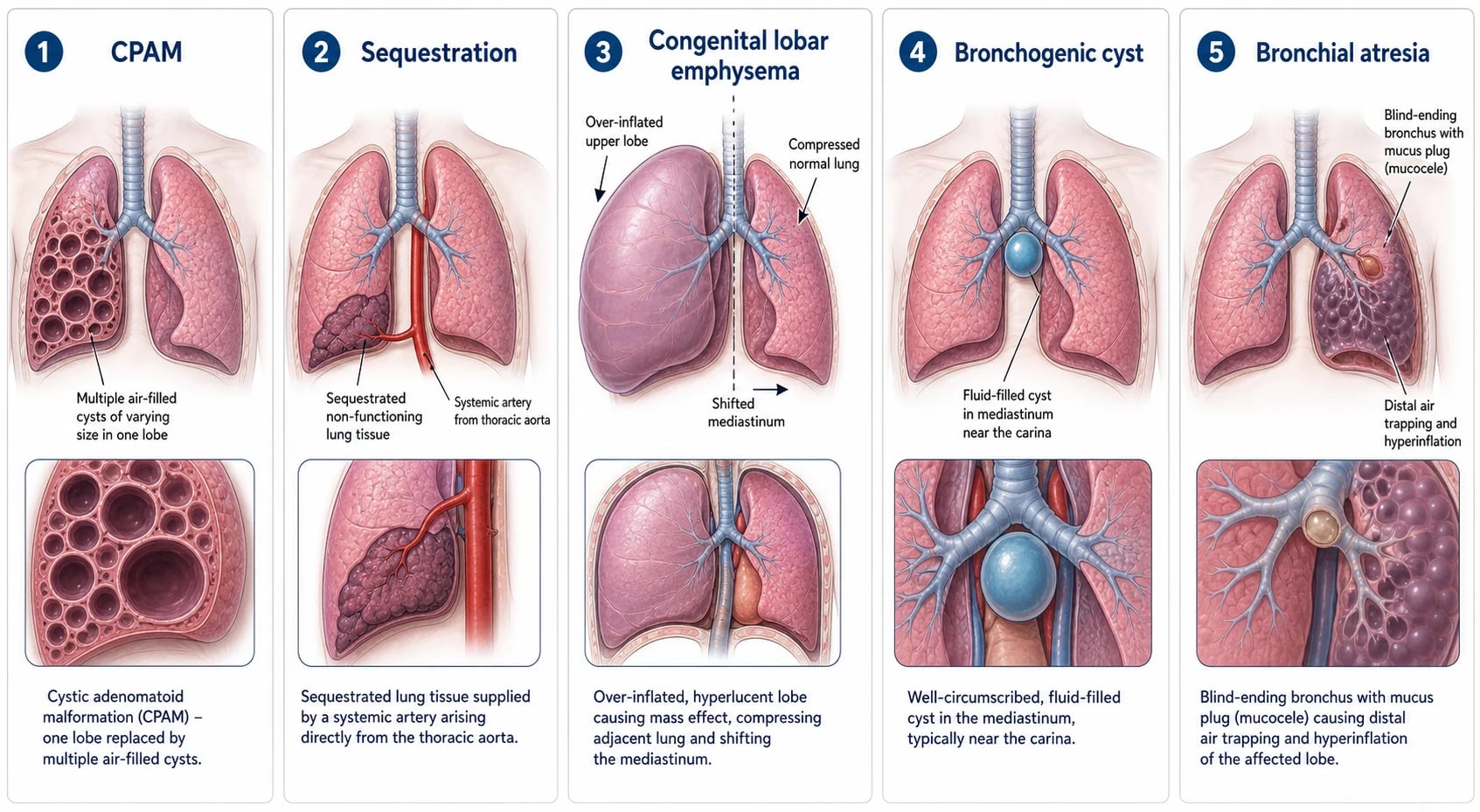
Among the pulmonary-supplied lesions, CPAM is the archetype — a mass of cystic and adenomatoid lung with disordered airway branching, graded by the Stocker system from the large-cyst type through microcystic and solid variants. Congenital lobar emphysema is a single lobe that progressively over-inflates behind a floppy or narrowed airway, a bronchogenic cyst is a fluid-filled foregut cyst usually sitting in the mediastinum near the carina, and bronchial atresia is a blind-ending bronchus with a mucus-filled stump and air trapping beyond it. [2] [1]
The airway malformations sit alongside this parenchymal spectrum and are dominated by obstruction of the large airways. The most dramatic is congenital high airway obstruction syndrome, where atresia or a web blocks the larynx or trachea, the lungs fill with trapped fluid, and the fetus develops massive lungs, a flattened diaphragm and hydrops — a picture that is now recognised antenatally and managed at specialist fetal centres. [10] [1]
Epidemiology & Risk Factors
These lesions are individually uncommon but collectively important, with congenital lung malformations detected in roughly one in two-and-a-half to one in eight thousand pregnancies depending on the population and the intensity of antenatal screening. CPAM and sequestration together account for the large majority, and improved ultrasound resolution has steadily lifted the detection rate over recent decades. [1] [8]
Most lesions are sporadic, with no clear inherited pattern and no strong maternal or environmental risk factor to counsel about, which is itself a reassuring message for families searching for something they did wrong. The important exception is the association between some cystic lesions and the DICER1 tumour-predisposition syndrome, where a family history of pleuropulmonary blastoma, cystic nephroma or thyroid disease changes the level of concern about a cystic lung lesion. [2] [6]
The strongest predictor of how a lesion behaves is not a risk factor at all but its antenatal size and physiology. A large lesion that shifts the mediastinum, compresses the developing lung or the heart, or provokes hydrops carries a very different outlook from the small lesion that plateaus and fades relative to the growing fetus, and gauging that size antenatally is what allows the fetal team to counsel and to intervene. [9] [8]
Pathophysiology
Each lesion is best understood as normal lung development thrown off course at a particular moment. The fetal lung grows by branching morphogenesis, dividing again and again from a single foregut bud through the embryonic, pseudoglandular, canalicular, saccular and alveolar stages, and an insult or a genetic misstep during this programme leaves a fixed structural fault in whichever region was forming at the time. [1] [2]
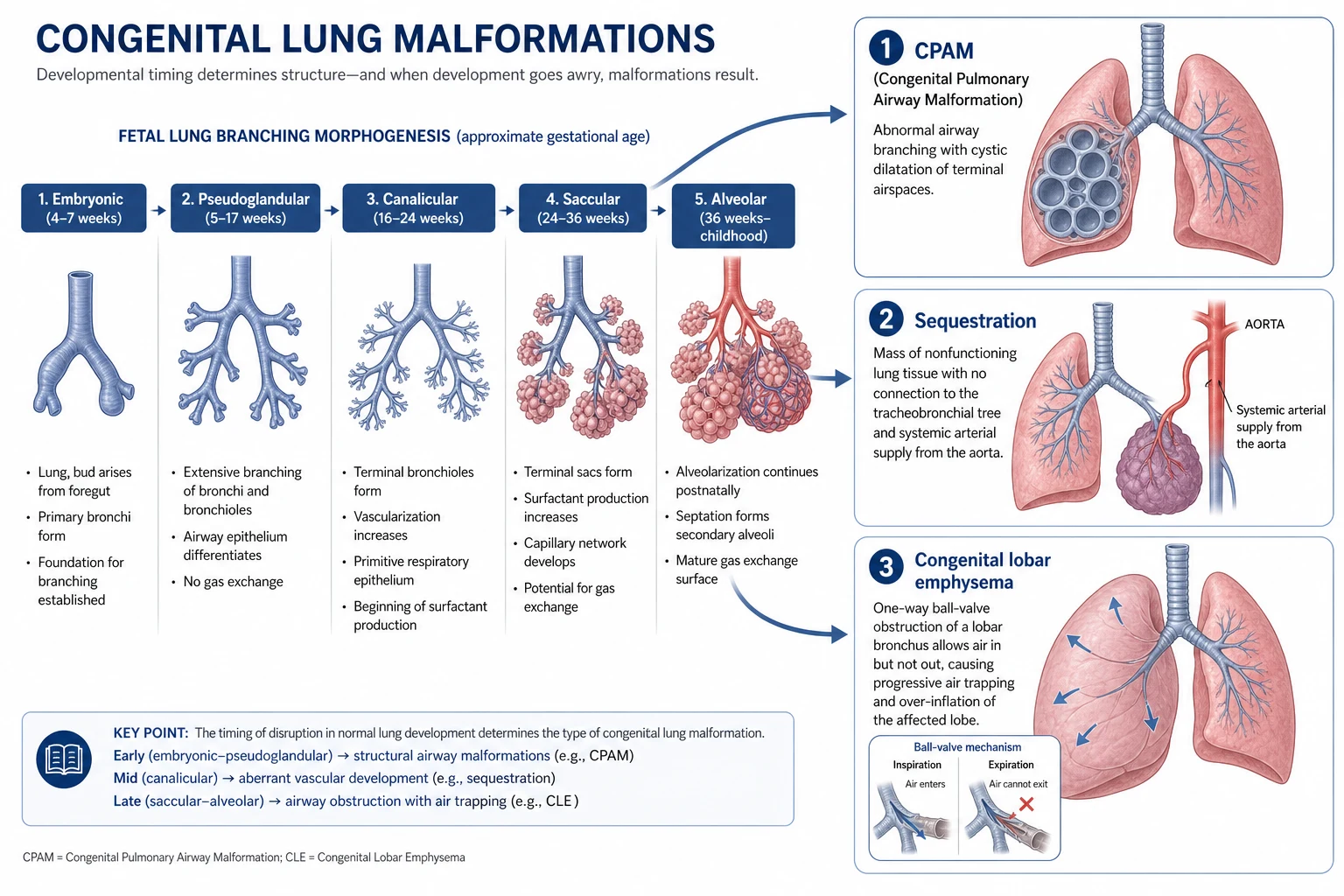
In CPAM the fault is disordered terminal airway branching, producing a mass of communicating cysts and adenomatoid tissue that connects, often abnormally, to the bronchial tree and so can trap air and become infected. In sequestration an accessory bud of lung tissue develops with no normal bronchial connection and recruits its own blood supply from a systemic artery, leaving a segment of non-functioning lung that is fed and drained outside the normal circuit. Congenital lobar emphysema is different again, driven by a one-way airway obstruction that lets a lobe fill but not empty, so it inflates relentlessly. [1] [11]
The antenatal natural history follows from this. Many lesions grow fastest in the mid-trimester, when the fetal lung is dividing most actively, then plateau or shrink relative to the enlarging fetus, which is why serial scanning matters and why an alarming early lesion can look far less threatening by term. When a lesion instead keeps growing, its mass compresses the heart and great veins, impairs venous return, and tips the fetus into hydrops — the physiological endpoint that fetal intervention is designed to prevent. [9] [7]
Clinical Presentation
The commonest presentation now is no presentation at all: a well, asymptomatic baby carried to term after an antenatal scan flagged a lesion, breathing normally and examining normally on the postnatal ward. Recognising that this is the usual picture keeps the clinician calm and focused on planned imaging rather than reflexive intervention, because the child who looks well genuinely is well in the short term. [8] [1]
At the other extreme, a large lesion declares itself as neonatal respiratory distress. A congenital lobar emphysema that keeps inflating, a big CPAM that fills a hemithorax, or a tension cyst can compress the normal lung and shift the mediastinum, producing tachypnoea, recession, hypoxia and, if unrelieved, cardiorespiratory compromise in the first hours to days of life. This is the presentation that turns an elective problem into an emergency. [1] [12]
Beyond the newborn period, a lesion that was missed or left in place tends to announce itself through infection. The classic story is pneumonia that keeps returning to the same lobe, sometimes with an abscess or empyema, because a poorly draining cystic or sequestered segment is an ideal reservoir for bacteria — and same-site recurrence is the signal to image the chest rather than prescribe again. [1] [3]
Differential Diagnosis
The single most important mimic to exclude on the antenatal scan is congenital diaphragmatic hernia, in which bowel loops in the chest can look strikingly like a cystic lung mass. Getting this wrong is dangerous, because a hernia demands a completely different delivery plan and immediate neonatal airway and gastric management, so the integrity of the diaphragm and the position of the stomach must be positively established before a lesion is filed under benign lung. [1] [8]
After birth, the postnatal differential of a cystic or hyperlucent area shifts toward acquired disease. A necrotising pneumonia with cavitation, a large pneumatocele, a simple pneumothorax and, importantly, the malignant pleuropulmonary blastoma can all resemble a congenital cystic lesion on plain imaging, and distinguishing them changes management entirely. The context — a well baby with an antenatal finding versus an acutely unwell child — usually points the way, but imaging settles it. [2] [6]
Because the malignant mimic is the one that punishes complacency, an atypical lesion deserves particular respect. A cystic lung lesion that behaves oddly, that clusters in a family with other DICER1-associated tumours, or that is resected and looks unusual under the microscope should prompt formal pathological review and genetic consideration rather than the assumption that every cyst is a benign CPAM. [2] [6]
Clinical & Bedside Assessment
Assessment begins before birth, in the fetal-medicine clinic, where serial ultrasound tracks the lesion's size and, crucially, its effect on the surrounding structures. The examiner wants to hear that you look not just at the lesion but at the mediastinum, the heart, the amniotic fluid and the fetal skin and abdomen for the early signs of hydrops, because it is the physiological consequences, not the raw dimensions, that determine risk. [9] [8]
At birth the examination of an asymptomatic baby is usually and reassuringly normal, and the skill is to document that normality carefully rather than to hunt for signs that are not there. Confirm a comfortable respiratory pattern, normal saturations, symmetrical air entry and no mediastinal shift, and take a focused history of the antenatal findings, the scans performed and any family history of DICER1-associated tumours. [8] [1]
In the older child who presents with infection rather than an antenatal diagnosis, the bedside task is the same discipline used for any recurrent pneumonia: verify the episodes with their films, establish whether they struck the same site, and examine for the fixed focal signs, clubbing or faltering growth that betray established suppurative disease behind an unrecognised malformation. Reviewing the actual radiographs is often the most revealing part of the assessment. [3] [1]
Investigations
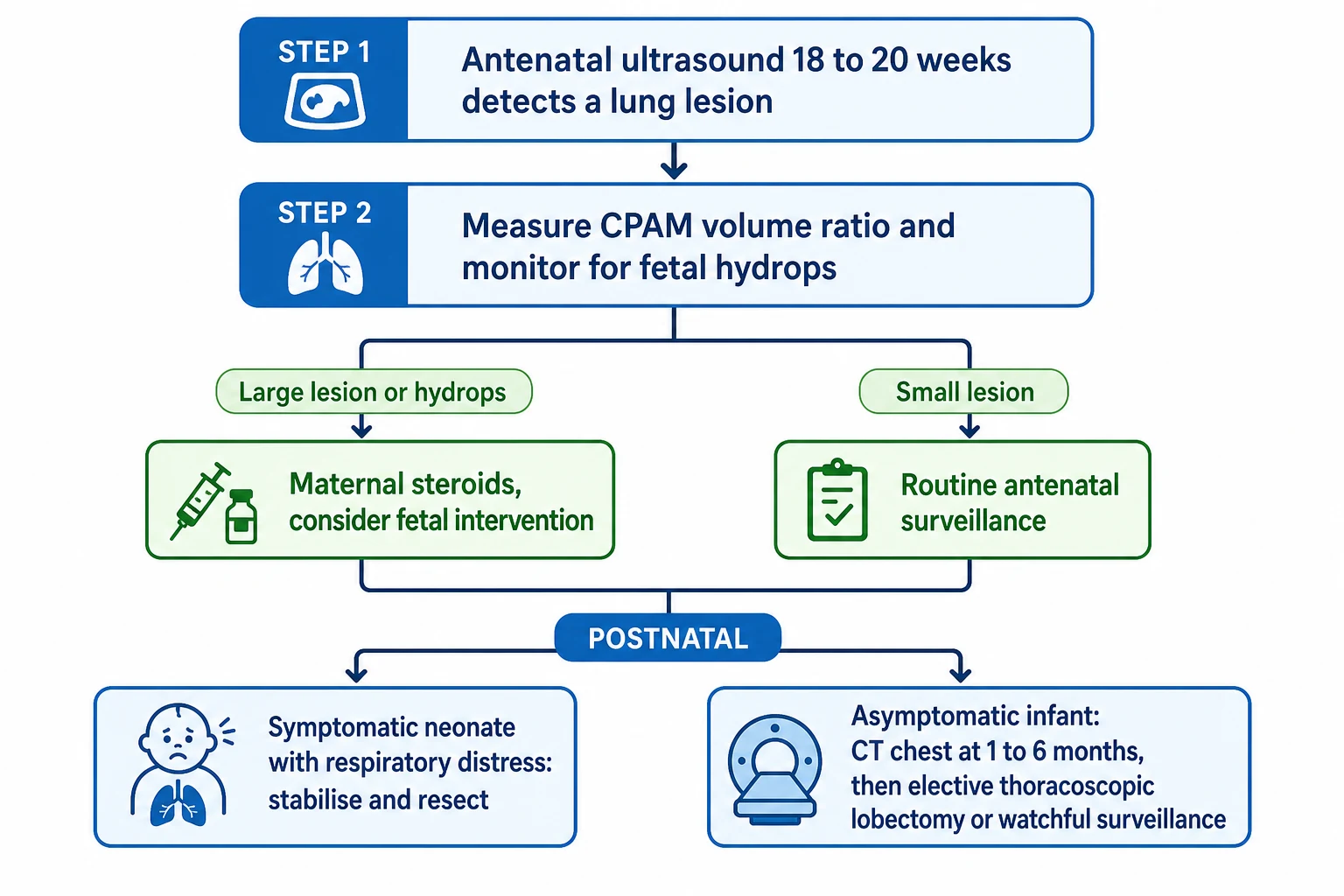
Antenatal imaging carries the early diagnostic weight. Detailed ultrasound characterises the lesion, identifies a systemic feeding artery that marks a sequestration, and is used to calculate the CPAM volume ratio, a size index that predicts the risk of hydrops and guides how closely the pregnancy is watched. Fetal MRI adds anatomical detail when the ultrasound is ambiguous or when planning delivery and surgery for a large lesion. [9] [8]
After birth the pivotal principle is that a normal chest radiograph does not exclude a malformation. Many lesions shrink or become inconspicuous on plain film as the neonatal lung fills with air, so a reassuring newborn X-ray must never be taken as proof that the antenatally detected lesion has vanished. The definitive postnatal investigation is a contrast-enhanced chest CT, which defines the lesion, maps any systemic feeding vessel, and detects lesions that were invisible on plain film. [1] [8]
The timing of the postnatal CT is a practical judgement rather than a fixed rule. An asymptomatic infant is usually imaged in the first few months of life, late enough to spare the smallest babies unnecessary anaesthesia and radiation yet early enough to characterise the lesion and inform the surgical decision, whereas a symptomatic baby is imaged and treated without delay. Matching the tempo of investigation to the child in front of you is the mark of a considered approach. [8] [4]
Management — Resuscitation
Most children with these lesions never need resuscitation, but the symptomatic neonate can be critically unwell and is managed on standard airway, breathing and circulation lines with one crucial modification. Because congenital lobar emphysema and tension cysts over-distend when pressurised, positive-pressure ventilation is applied cautiously, the aim is to avoid worsening the air trapping, and definitive relief comes from urgent surgical decompression or resection rather than from escalating ventilator settings. [1] [12]
The fetal emergency is the large lesion driving hydrops, and here the intervention happens before birth. A course of maternal corticosteroid can shrink a large microcystic CPAM and reverse early hydrops, a dominant macrocyst can be drained or shunted, and the largest hydropic lesions may be considered for open fetal surgery at specialist centres — all decisions made by a fetal-medicine team once the volume ratio and hydrops mark the fetus as high risk. [7] [9]
For congenital high airway obstruction syndrome, the resuscitation challenge is the airway itself at the moment of birth, and the solution is to secure it while placental circulation is preserved through an ex-utero intrapartum treatment procedure, a planned delivery designed to buy time to establish an airway before the cord is clamped. This is high-level, centralised care with survival now reported in a meaningful proportion of these once-uniformly-fatal cases. [10] [1]
Management — Definitive & Stepwise
Definitive management divides cleanly along the symptomatic line. A symptomatic lesion — one causing respiratory distress, recurrent infection or a complication — is resected, and resection reliably relieves symptoms and removes the reservoir for future infection and malignant change. The standard operation is lobectomy, increasingly performed thoracoscopically in experienced hands, with lung-sparing segmentectomy an option that preserves parenchyma when the anatomy allows. [5] [6]
The genuine controversy is the asymptomatic, antenatally detected lesion, where thoughtful clinicians disagree. The case for elective resection rests on removing a lifelong reservoir for infection and a small but real risk of malignancy, and on the technical ease of operating on a well infant; the case for watchful surveillance rests on the fact that many lesions never cause trouble and on the wish to avoid an operation the child might never have needed. Recent syntheses suggest conservative management of a truly asymptomatic CPAM can be safe with structured follow-up, while other cohorts still favour early surgery, and the honest examination answer is to lay out both sides. [3] [4]
Stepwise approach to a congenital lung malformation
Confirm the antenatal diagnosis and exclude a congenital diaphragmatic hernia by checking the diaphragm and stomach
Track lesion size and hydrops risk with serial ultrasound and the CPAM volume ratio
Intervene before birth for the hydropic fetus with maternal steroids, drainage or specialist fetal surgery
Assess the newborn: stabilise and resect the symptomatic baby, avoiding over-distension of air-trapping lesions
Obtain a timed contrast-enhanced chest CT in the asymptomatic infant even when the plain film looks normal
Decide with the family between elective resection and structured surveillance for the asymptomatic lesion
When surgery is chosen for a well child, it is usually planned electively in the first year of life, balancing the technical and immunological advantages of operating early against anaesthetic considerations in the smallest infants. Where surveillance is chosen, it must be genuinely structured — clear imaging intervals, defined triggers to operate, and a family who understand the plan — rather than a euphemism for losing the child to follow-up. [4] [3]
CLASH — the congenital lung malformation spectrum
Specific Subtypes & Scenarios
CPAM is the subtype to master because it dominates the spectrum and carries the malignancy question. The Stocker grading spans a large-cyst type, a common intermediate type, and a solid microcystic type, and the important clinical point is that some cystic lesions overlap pathologically with type 1 pleuropulmonary blastoma, which is why an atypical CPAM or a DICER1 family history changes the threshold for resection and pathological scrutiny. [2] [6]
Bronchopulmonary sequestration is defined by its rogue blood supply and comes in two forms: an intralobar sequestration sharing the visceral pleura of a normal lobe and presenting later with infection, and an extralobar sequestration with its own pleural covering, often found antenatally and sometimes below the diaphragm. Identifying the systemic feeding artery is essential, both to confirm the diagnosis and to warn the surgeon or interventional radiologist, since some lesions are amenable to arterial embolisation. [11] [1]
Congenital lobar emphysema and the bronchogenic cyst round out the high-yield scenarios. The lobar emphysema is the neonatal-distress lesion whose management hinges on not over-pressurising it and on timely lobectomy, while the bronchogenic cyst is a mediastinal foregut cyst that may be silent for years or may compress the airway or oesophagus, and a symptomatic or enlarging cyst is resected. Both illustrate the same rule that symptoms drive intervention. [12] [1]
Complications & Pitfalls
The complications that matter are infection, mechanical compromise and malignancy. A poorly draining lesion becomes a nidus for recurrent same-site pneumonia, abscess and empyema; a large or air-trapping lesion compresses normal lung and the mediastinum; and, uncommonly but importantly, a cystic lesion can harbour or evolve into pleuropulmonary blastoma or, later, an adenocarcinoma, which is the argument that keeps resection on the table even for the well child. [6] [3]
The commonest cognitive pitfall is false reassurance. A well baby and a clean-looking chest film tempt the team to close the file, yet the lesion may simply be inconspicuous on plain imaging and the child may return years later with infection or, rarely, a tumour. The corrective discipline is to complete the planned CT, make a deliberate decision about surgery versus surveillance, and hand the family a clear plan rather than a vague promise to keep an eye on things. [1] [4]
Prognosis & Disposition
The overall outlook is good, and this is the reassuring headline for families. A symptomatic lesion that is resected usually resolves completely, the remaining lung compensates well in early childhood, and long-term respiratory function after lobectomy in infancy is generally preserved, so a child who needs an operation can be expected to grow up with normal exercise tolerance in most cases. [5] [4]
The prognosis for the asymptomatic lesion is the crux of the surveillance debate rather than a settled fact. Long-term comparative data suggest that carefully selected asymptomatic lesions can be watched without harm, while other cohorts report infection or the discovery of malignancy over time, and the truthful position is that both operative and conservative pathways can deliver good outcomes provided follow-up is reliable. What predicts a poor outcome is not the pathway but a lesion lost to follow-up or a complication missed. [4] [3]
Disposition is therefore about durable, coordinated follow-up. Every child needs a definitive postnatal diagnosis, an explicit decision about surgery or surveillance, and, where the lesion is watched, a named plan with imaging intervals and triggers to operate. Children treated at fetal or paediatric surgical centres should be transitioned back to shared local care with that plan documented, so the thread is never dropped between services. [4] [1]
Special Populations
The fetus is the first special population, and the antenatally diagnosed hydropic lesion is the group in whom timing and centralisation change survival. A fetus with a large lesion, mediastinal compression and early hydrops needs referral to a fetal-medicine centre capable of steroids, shunting, fetal surgery and, for airway atresia, an EXIT delivery, because these interventions are only available in a handful of specialist units and delay costs lives. [7] [10]
Children with complex, technology-dependent or syndromic backgrounds carry added risk and complexity. A congenital lung lesion in a child with other anomalies, chronic ventilation needs or a tumour-predisposition syndrome demands multidisciplinary planning, careful anaesthetic assessment, and a lower threshold for genetic evaluation, since the malformation rarely exists in isolation and its management must fit the whole child. [6] [2]
Access and equity form the third dimension. Families in rural, remote and socioeconomically disadvantaged communities, including Aboriginal and Torres Strait Islander families, may live far from the fetal-medicine and paediatric surgical centres where this care is concentrated, and a lesion that recurs with infection is more harmful when specialist review and definitive imaging are hard to reach. Deliberate coordination, telehealth and culturally safe pathways help keep these children within the plan. [1] [3]
Evidence, Guidelines & Regional Differences
The modern evidence base is anchored by comprehensive reviews that frame congenital lung malformations as one overlapping spectrum with shared antenatal and surgical principles, and by pathological reconsiderations of the Stocker classification that keep the malignancy link in view. These sources set the conceptual map on which the management debates are argued. [1] [2]
Management of the individual limbs rests on more focused work. Trials and cohorts on maternal betamethasone underpin antenatal steroid use for large microcystic lesions and hydrops, volume-ratio studies guide antenatal risk stratification, surgical series and meta-analyses inform the choice between lobectomy and segmentectomy, and dedicated fetal-centre reports document the emerging survival in congenital high airway obstruction syndrome. [7] [5]
The central controversy remains the asymptomatic lesion, and the literature genuinely diverges. Systematic reviews increasingly support the safety of structured conservative management of a truly asymptomatic CPAM, while propensity-matched cohorts continue to weigh the long-term balance of operative against conservative care, and the smaller unresolved questions concern the optimal timing of elective surgery and the choice of lobectomy versus lung-sparing resection. The examiner wants a candidate who can hold both sides honestly. [3] [4]
Exam Pearls
The spine of the topic is the antenatal transformation and the two organising questions. Most lesions are found before birth in a well fetus, so the first question is whether the lesion is causing trouble now — hydrops before birth or distress after it — because that child is stabilised and resected. The second and harder question is what to do with the far commoner asymptomatic lesion, and a strong answer lays out the resection-versus-surveillance debate rather than pretending it is settled. [1] [3]
The set-piece facts follow from the anatomy. Learn the spectrum through a simple scheme, remember that a systemic feeding artery defines sequestration, and hold the safety instincts that examiners reward: a normal newborn film never excludes a lesion, a hyperlucent neonatal thorax may be lobar emphysema that must not be over-pressurised, and an antenatal chest mass could be a diaphragmatic hernia until the diaphragm is confirmed. [8] [12]
Finally, show that you understand why the debate matters. The reason clinicians still resect well children is the reservoir for recurrent infection and the small but real link between cystic lesions and pleuropulmonary blastoma, and the reason others watch is that many lesions never cause harm and surgery carries its own cost. Demonstrating that you can weigh malignancy risk, infection risk and operative risk together is what marks the candidate who truly understands congenital lung malformations. [6] [4]
References
- [1]Pederiva F, Rothenberg SS, Hall N, et al Congenital lung malformations. Nat Rev Dis Primers, 2023.PMID 37919294
- [2]Dehner LP, Schultz KAP, Hill DA Congenital Pulmonary Airway Malformations With a Reconsideration and Current Perspective on the Stocker Classification. Pediatr Dev Pathol, 2023.PMID 37334833
- [3]Thorburn C, Kattini C, El Hafid M, et al The safety of conservative management of asymptomatic congenital pulmonary airway malformations (CPAMs) in children: A systematic review. J Pediatr Surg, 2026.PMID 41643769
- [4]Lambrecht S, Elrod J, Thater G, et al Long-term outcomes after operative versus conservative management of congenital thoracic malformations: a propensity-matched cohort study. Pediatr Surg Int, 2026.PMID 42043561
- [5]Mohamed SF, Abouegla M, Abouzeid M, et al Efficacy of lobectomy versus segmentectomy for congenital lung malformations: a systematic review and meta-analysis. Pediatr Surg Int, 2026.PMID 41879907
- [6]Muntean A, Marsland L, Sikdar O, et al Neonatal Surgery for Congenital Lung Malformations: Indications, Outcomes and Association With Malignancy. J Pediatr Surg, 2025.PMID 40031114
- [7]Peranteau WH, Boelig MM, Khalek N, et al Effect of single and multiple courses of maternal betamethasone on prenatal congenital lung lesion growth and fetal survival. J Pediatr Surg, 2016.PMID 26526208
- [8]Bush A Prenatal presentation and postnatal management of congenital thoracic malformations. Early Hum Dev, 2009.PMID 19758773
- [9]Kontopoulos E, Quintero LF, Quintero Kontopoulos A, et al Mathematical reappraisal of the congenital cystic adenomatoid malformation volume ratio. J Matern Fetal Neonatal Med, 2026.PMID 42010266
- [10]Wagner ML, Peiro JL, Rymeski BA, et al Long-term outcomes of congenital high airway obstruction syndrome (CHAOS) at a single comprehensive fetal center. Fetal Diagn Ther, 2026.PMID 42213638
- [11]Hong F, Cai W, Li J, et al Pulmonary sequestration with a rare renal artery subtype: a comprehensive clinicopathological analysis of 20 pediatric cases. BMC Pulm Med, 2026.PMID 42046053
- [12]Abboud F, Muhammad AA, Ali S, et al Congenital Lobar Emphysema in an Infant Presenting With Persistent Cough and Progressive Respiratory Symptoms: A Case Report. Clin Case Rep, 2026.PMID 42063629