Paeds · respiratory-sleep-and-airway
Interstitial lung disease in children
Also known as chILD · Children's interstitial lung disease · Childhood interstitial and diffuse lung disease · Diffuse lung disease in children · Paediatric interstitial lung disease
Fellowship guide to children's interstitial lung disease (chILD) — the chILD syndrome that flags it, the infancy-versus-older-child classification that frames it, the surfactant-dysfunction and neuroendocrine-cell-hyperplasia biology that drives the commonest genetic forms, the HRCT-genetics-biopsy pathway that names it, and the supportive-plus-disease-specific management delivered through a specialist chILD centre.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
An infant of a few months is breathing fast all the time, tires during feeds, is slipping down the weight centiles, and has fine crackles heard across both lungs. Bronchiolitis was diagnosed twice, but the tachypnoea never really settled and the oxygen saturations sit a little low. The reflex to resist is another round of antibiotics; the thought to have instead is that a persistently abnormal interstitium can look exactly like this, and that this baby may have children's interstitial lung disease. [1] [3]
Children's interstitial lung disease, universally abbreviated to chILD, is not one disease but an umbrella term for more than a hundred rare disorders that share a common target: the alveolar-interstitial region of the lung. Whatever the trigger, the interstitium and the surrounding alveoli become thickened, inflamed, or scarred, so the delicate barrier for gas exchange no longer works efficiently. The result is chronic hypoxaemia and breathlessness out of proportion to any single infection. [3] [7]
Because the individual disorders are rare and easily missed, the field agreed on a practical entry point called the chILD syndrome. A child is said to have the chILD syndrome when diffuse lung disease is present with at least three of four features — respiratory symptoms such as cough or exertional breathlessness, respiratory signs such as tachypnoea, crackles, retractions or clubbing, hypoxaemia, and diffuse abnormalities on a chest radiograph or CT — and the more common explanations have been excluded. [1] [4]
That definition is deliberately a screening trigger rather than a final answer. Meeting the chILD syndrome does not name the disease; it says the child has crossed the threshold that demands a structured diffuse-lung-disease evaluation. The job that follows is to exclude the everyday causes, then work through imaging, genetics, and sometimes biopsy to reach the specific diagnosis that decides treatment and prognosis. [1] [4]
Classification
The most useful way to hold this enormous list in mind is to split it by the age at which the disorders appear, because the biology and the work-up differ sharply between the two groups. One group is largely unique to infancy and is dominated by problems of lung growth and surfactant; the other affects older children and resembles the interstitial lung diseases seen in adults. This age split is the backbone of the modern classification. [2] [1]
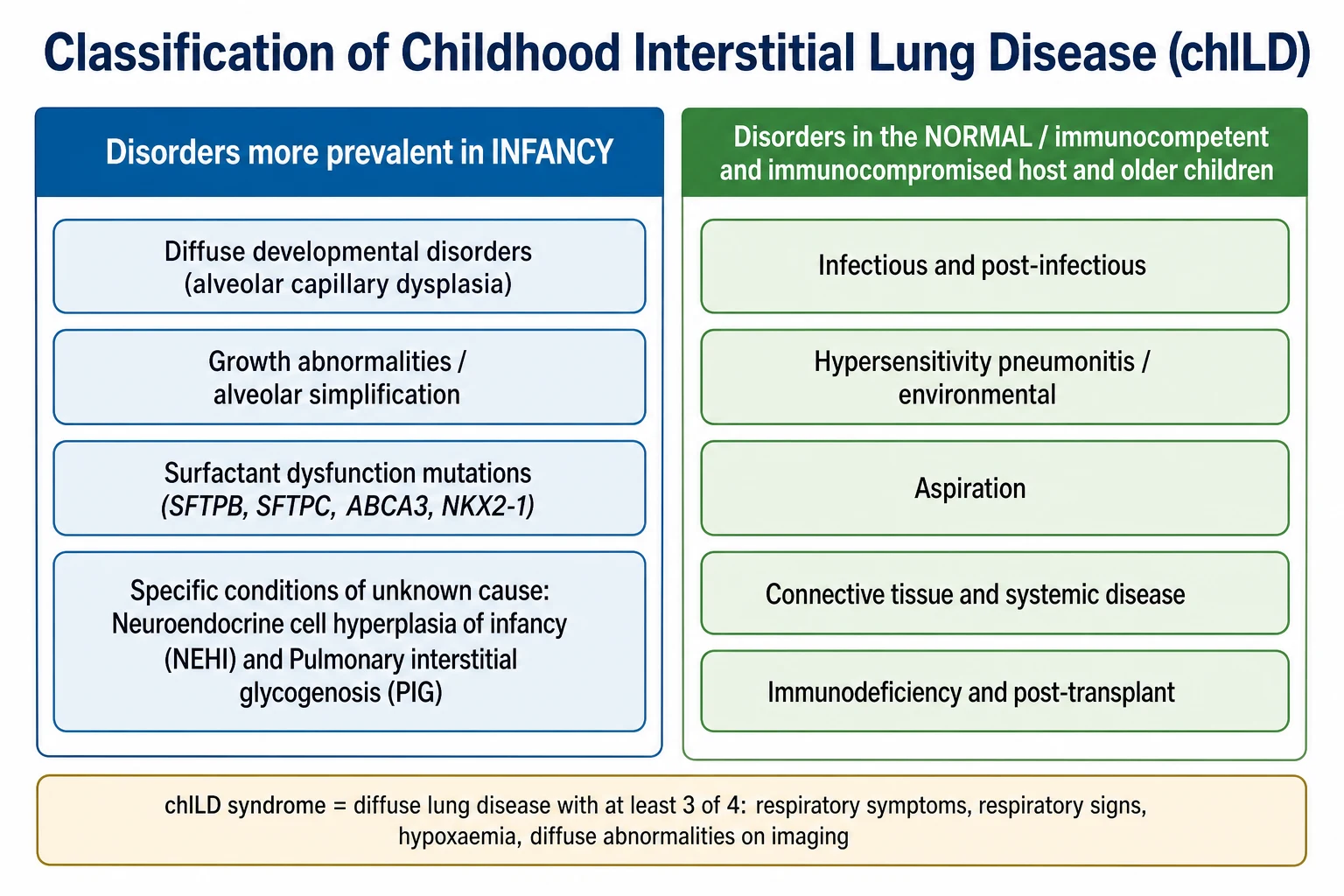
The infancy-predominant group is the one to know in detail for paediatric practice. It contains diffuse developmental disorders such as alveolar capillary dysplasia, growth abnormalities with alveolar simplification seen in chronic neonatal lung disease, the genetic surfactant dysfunction disorders, and the specific conditions of unknown cause — above all neuroendocrine cell hyperplasia of infancy and pulmonary interstitial glycogenosis. These are the entities that a general paediatrician must recognise as distinctly paediatric. [2] [1]
The older-child group is organised by host and exposure rather than by development. It covers infectious and post-infectious disease, hypersensitivity pneumonitis and other environmental exposures, aspiration, the interstitial disease of connective tissue and systemic disorders, and the lung disease of immunodeficiency or transplantation. In this group the reasoning mirrors adult interstitial lung disease, and the search focuses on immune, systemic, and exposure causes. [3] [8]
Epidemiology & Risk Factors
Children's interstitial lung disease is genuinely rare, and that rarity is its central clinical problem. Population estimates put chILD at roughly one to a few cases per million children, so a general paediatrician may see only a handful in a career, and the disorders are frequently mislabelled as recurrent infection, difficult asthma, or bronchiolitis that will not resolve. The commonest reason the diagnosis is missed is simply that it is not considered. [3] [7]
Within that rarity, age and genetics shape the risk. The surfactant dysfunction disorders are inherited, so a family history of neonatal respiratory death, of chronic lung disease across generations, or of parental consanguinity raises the odds of a genetic surfactant defect. Neuroendocrine cell hyperplasia of infancy characteristically appears in the first year or two of life in an otherwise thriving infant, and some familial clustering has been described. [10] [5]
Prematurity and its aftermath contribute through a different route. The growth abnormalities with alveolar simplification overlap with chronic neonatal lung disease, so ex-preterm infants and those with syndromes affecting lung growth are over-represented in the developmental and growth groups. In older children, immunodeficiency, connective tissue disease, transplantation, and environmental or drug exposures are the principal acquired risk factors. [2] [8]
Pathophysiology
Every disorder in this group converges on one final problem: the wall between air and blood stops working. In health the alveolar-interstitial region is exquisitely thin, letting oxygen and carbon dioxide diffuse freely. In chILD that region becomes thickened by cellular infiltrate, abnormal storage material, poorly formed alveoli, or fibrosis, so gas exchange falls and the child compensates by breathing faster. Understanding this shared endpoint explains why such different diseases present so similarly. [3] [7]
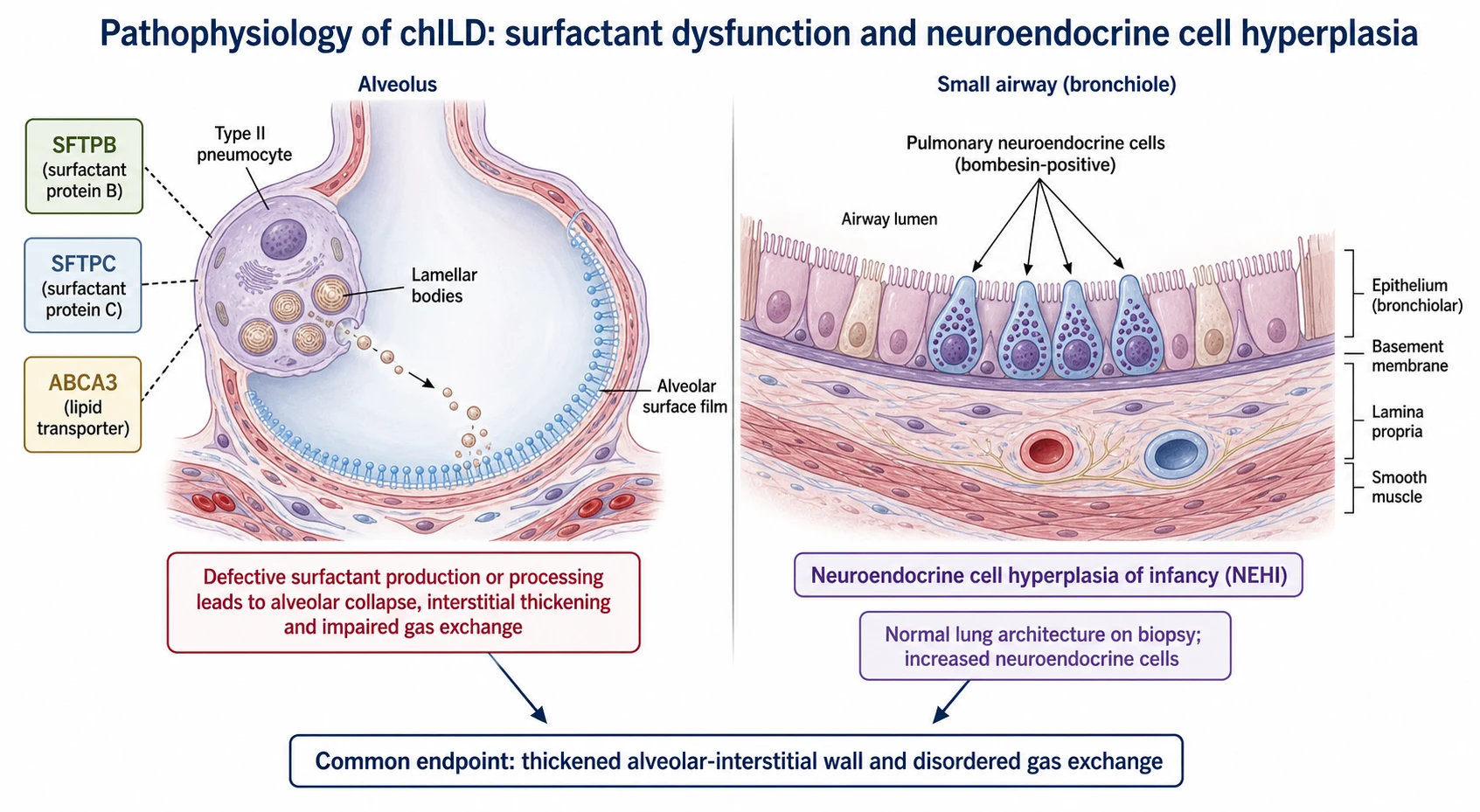
The surfactant dysfunction disorders are the mechanistic centrepiece and the reason genetics has transformed the field. Surfactant is made and processed by the type II alveolar pneumocyte, which packages it into lamellar bodies and secretes it to keep alveoli open. A defect in surfactant protein B or the lipid transporter ABCA3 disables that process from birth and typically causes severe, often lethal, neonatal disease, whereas a surfactant protein C mutation more often produces chronic, variable interstitial disease across childhood. [10] [11] [12]
Neuroendocrine cell hyperplasia of infancy works by a strikingly different mechanism and is the counterpoint to remember. Here the lung architecture is essentially normal on biopsy, but there is a marked increase in bombesin-positive pulmonary neuroendocrine cells within the small airways. This cellular excess, rather than any interstitial scarring, is thought to drive the persistent tachypnoea and hypoxaemia, which is why NEHI behaves so much more benignly than the surfactant disorders. [5] [6]
The remaining groups reach the same functional endpoint by their own routes. Growth abnormalities leave the lung with too few, oversimplified alveoli; developmental disorders such as alveolar capillary dysplasia misplace the pulmonary vessels so blood and air never meet properly; and the older-child diseases thicken the interstitium through immune inflammation, aspiration injury, or fibrosis. In each case the mechanism differs but the physiological cost — impaired gas exchange in a diffusely abnormal lung — is shared. [2] [8]
Clinical Presentation
The presentation that should trigger the whole approach is chronic, unexplained respiratory distress in a young child, and the signature is tachypnoea that persists between and beyond acute illnesses. The infant breathes fast at rest, works harder than expected with subcostal and intercostal recession, tires during feeds, and often fails to thrive because the extra work of breathing burns energy. This is a child who is chronically breathless, not one who is intermittently wheezy. [1] [3]
On examination the most useful sign is diffuse fine inspiratory crackles heard across both lung fields, which point to the interstitium rather than to the large airways. Hypoxaemia is common and may be subtle at rest but unmasked by feeding, sleep, or exertion, so oximetry in these states is revealing. Digital clubbing develops in more chronic or severe disease and is a marker that the process has been present and significant for some time. [1] [7]
The tempo and the starting age help sort the differential. A term newborn with severe, progressive, and treatment-resistant respiratory failure suggests a lethal developmental or surfactant disorder such as surfactant protein B or ABCA3 deficiency. An otherwise well-grown infant with persistent tachypnoea, crackles, and mild hypoxaemia over months, appearing after the first weeks of life, fits neuroendocrine cell hyperplasia of infancy. An older child with progressive exertional breathlessness and a systemic illness points to the host-based group. [5] [11]
Differential Diagnosis
The first and most important differential task is to exclude the common causes, because the chILD syndrome is only meaningful once they are gone. Cystic fibrosis, primary and secondary immunodeficiency, recurrent aspiration, congenital heart disease with pulmonary oedema, and chronic infection can all produce diffuse lung change and breathlessness in a young child, and each is far more common than any chILD disorder. Working through them is the mandatory first pass. [1] [4]
Only when those everyday explanations are excluded does the diffuse change earn a chILD label, and the differential then narrows by age and clues. In infancy the question becomes which of the developmental, growth, surfactant, or NEHI disorders fits the tempo, the family history, and the imaging. In older children the search shifts to hypersensitivity pneumonitis, aspiration, connective tissue disease, and immunodeficiency-related lung disease, echoing the reasoning of adult ILD. [2] [8]
The trap to avoid is the mirror image of over-calling and under-calling. Labelling a genuinely diffuse, persistent interstitial process as yet another chest infection delays a treatable or genetically important diagnosis; equally, jumping to an invasive chILD work-up before excluding cystic fibrosis or immunodeficiency wastes time and risk. The discipline is to exclude the common first, then pursue the rare methodically. [1] [4]
Clinical & Bedside Assessment
The history is where the diagnosis is usually first suspected, and it has three tasks: establish that the respiratory trouble is chronic and diffuse, gauge its severity, and hunt for pointers to a specific cause. Ask how long the fast breathing has been present, whether it ever fully settles, how feeding and growth are going, and whether oxygen has ever been needed. A detailed family history of neonatal deaths, childhood lung disease, and consanguinity is essential when a genetic surfactant disorder is possible. [1] [4]
The examination is a deliberate search for the fingerprints of diffuse lung disease and its complications. Count the respiratory rate at rest, look for recession and the effort of breathing, and listen for diffuse fine crackles rather than focal or wheezy signs. Plot growth carefully, inspect for digital clubbing, and assess oxygenation with pulse oximetry not only at rest but during feeding, sleep, and activity, because desaturation is often positional and effort-dependent. [1] [7]
Beyond the general assessment, look actively for signs of severity and of an underlying systemic disorder. Signs of pulmonary hypertension, right heart strain, or worsening oxygen requirement mark severe disease that needs urgent specialist involvement. In the older child, examine the skin, joints, and other systems for the features of connective tissue disease or immunodeficiency, because the lung may be the presenting face of a systemic illness. [8] [3]
Investigations
Investigation runs on two tracks at once: exclude the common causes and characterise the diffuse lung disease. The exclusion track includes a sweat test and cystic fibrosis genetics, an immune screen, an assessment of swallowing and aspiration risk, and an echocardiogram to exclude congenital heart disease and to look for pulmonary hypertension. Only when this track is clear does the diffuse change confidently become chILD. [1] [4]
The characterising track is led by high-resolution CT of the chest, which is far more informative than a plain radiograph and often suggests the diagnosis. Certain HRCT patterns are close to diagnostic: the combination of ground-glass opacity in the right middle lobe and lingula with air trapping is characteristic of neuroendocrine cell hyperplasia of infancy and can, in the right clinical setting, spare the child a biopsy. Controlled-ventilation or age-appropriate technique improves image quality in infants. [6] [4]
Genetic testing has moved to the front of the pathway and frequently makes the diagnosis without surgery. A panel covering the surfactant genes — surfactant protein B, surfactant protein C, ABCA3, and NKX2-1 — should be sent early in an infant with diffuse lung disease, because a positive result confirms the diagnosis, guides prognosis and counselling, and can avoid an invasive biopsy. Infant lung function testing and airway sampling add supporting information in specialist centres. [11] [10]
Lung biopsy remains the reference standard when imaging and genetics leave the diagnosis unresolved, and it should be a video-assisted thoracoscopic biopsy handled by a centre with paediatric ILD pathology expertise. The tissue is examined with the specific stains and techniques the field has standardised, including immunostaining for neuroendocrine cells when NEHI is considered. Biopsy is targeted at the child in whom the answer will change management, not fired off routinely. [2] [1]
Management — Resuscitation
Although chILD is a chronic disease, children can present acutely and unstable, and the first task is always to stabilise the physiology in front of you. A child with severe hypoxaemia or respiratory failure needs supplemental oxygen titrated to maintain adequate saturations, careful support of work of breathing, and escalation to high-flow, non-invasive, or invasive ventilation when required. An intercurrent infection is a common precipitant and is treated promptly alongside the supportive measures. [1] [4]
The neonatal emergency is the term newborn with severe, progressive, treatment-resistant respiratory failure and diffuse lung disease, in whom a lethal surfactant or developmental disorder must be considered. These infants may need full intensive care support including, in selected cases, extracorporeal membrane oxygenation as a bridge while urgent genetic testing clarifies the diagnosis and the prognosis. Early genetic results are decisive because they shape the intensity of ongoing support and the conversation with the family. [11] [12]
Pulmonary hypertension is the complication that most often turns chronic chILD into an acute emergency, and it demands recognition and specialist management. A rising oxygen requirement, signs of right heart strain, or syncope should trigger echocardiography and urgent referral, because untreated pulmonary hypertension is a major driver of mortality in these children. Stabilise oxygenation, treat any precipitant, and involve the specialist centre without delay. [7] [1]
Management — Definitive & Stepwise
Definitive management rests on two pillars: excellent supportive care for every child, and disease-specific therapy for those who need and will benefit from it, all coordinated through a specialist chILD centre. Supportive care is the foundation and is often what changes the trajectory: supplemental oxygen to correct hypoxaemia and protect growth and the pulmonary circulation, aggressive nutritional support to overcome the high work of breathing, complete immunisation including influenza, respiratory syncytial virus prophylaxis for eligible infants, and strict avoidance of tobacco smoke. [1] [4]
Disease-specific pharmacotherapy is reserved for the disorders that have an inflammatory component or a demonstrated response, and it is not a blanket treatment for the whole spectrum. Systemic corticosteroids, given as pulses or as a continuous course, are the mainstay of anti-inflammatory treatment in many of the more inflammatory chILD disorders, sometimes with steroid-sparing agents such as hydroxychloroquine or azithromycin added in selected children. The evidence base is limited and largely observational, so treatment is individualised in a specialist setting. [7] [4]
Prednisolone / methylprednisolone (anti-inflammatory chILD, specialist-directed)
Dose
Commonly oral prednisolone about 1 to 2 mg/kg/day, or intravenous methylprednisolone pulses of about 10 to 30 mg/kg/day for three consecutive days repeated monthly, according to the specific disorder and centre protocol
Hydroxychloroquine is the additional agent used most often as a corticosteroid-sparing option, particularly in surfactant protein C disease and some other chronic inflammatory forms. It is added and monitored within the specialist team, with attention to its ophthalmological safety profile, and like the corticosteroids its use rests on observational experience rather than randomised trials. The overarching principle is that pharmacotherapy is matched to the specific diagnosis, not applied to chILD as a category. [7] [4]
Hydroxychloroquine (steroid-sparing, selected chronic chILD)
Dose
Commonly about 6 to 10 mg/kg/day orally in one or two divided doses, within specialist chILD centre protocols
For the most severe and progressive disease — above all the lethal surfactant protein B and ABCA3 deficiencies — lung transplantation is the only definitive treatment and must be considered early while the child is a viable candidate. At the other extreme, neuroendocrine cell hyperplasia of infancy needs no immunosuppression at all: it is managed with supportive oxygen and nutrition and tends to improve gradually over years. Matching the intensity of treatment to the specific diagnosis is the central management skill. [11] [5]
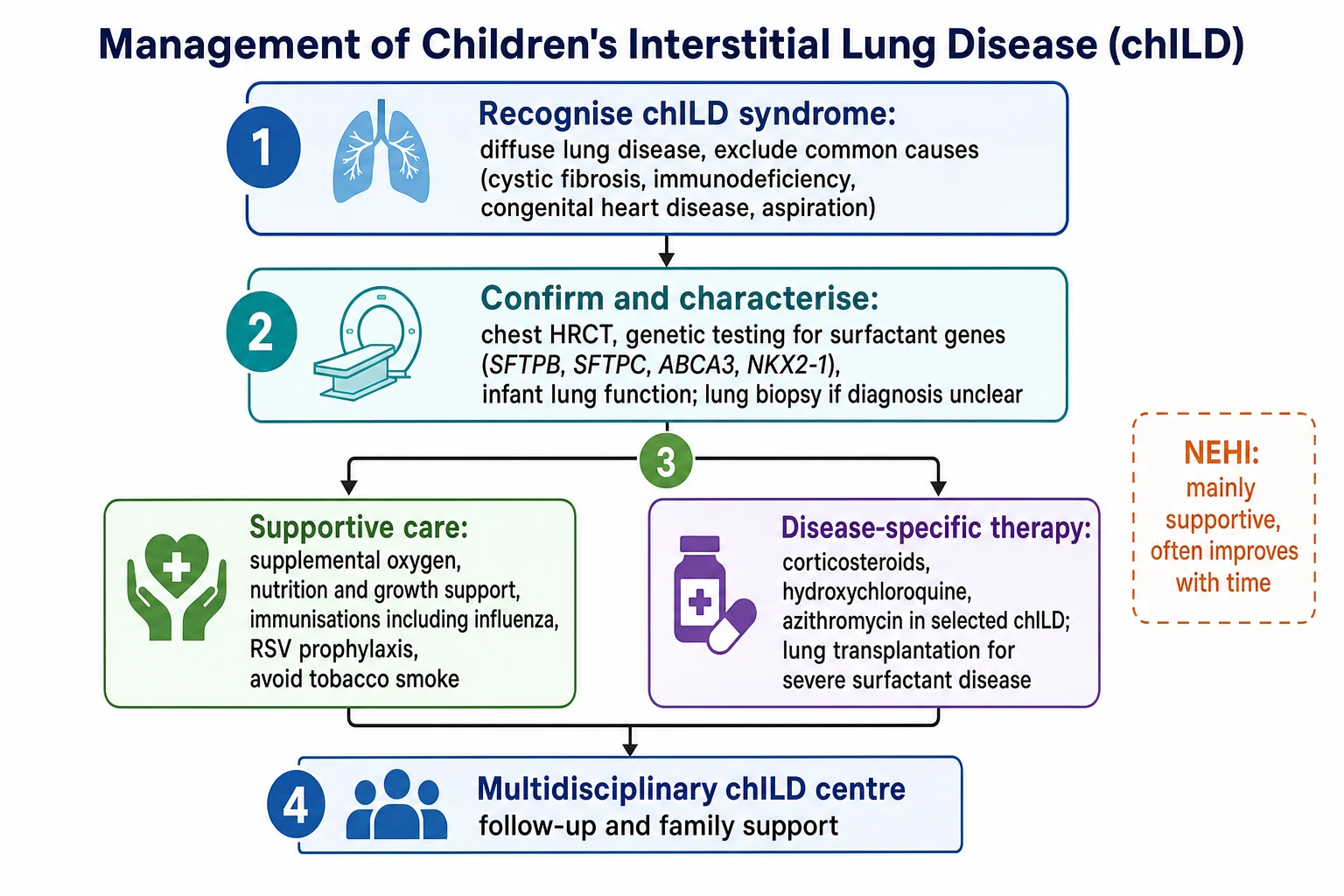
Stepwise approach to chILD
Recognise the chILD syndrome: diffuse lung disease with at least three of four features once common causes are considered
Exclude the common mimics: cystic fibrosis, immunodeficiency, congenital heart disease, and aspiration
Characterise with chest HRCT, which can be near-diagnostic for neuroendocrine cell hyperplasia of infancy
Send an early surfactant-gene panel (SFTPB, SFTPC, ABCA3, NKX2-1) in infants to confirm and to avoid biopsy where possible
Reserve video-assisted lung biopsy for cases unresolved by imaging and genetics
Give supportive care to every child: oxygen, nutrition, immunisation, RSV prophylaxis, smoke avoidance
Add disease-specific therapy (corticosteroids, hydroxychloroquine) only for inflammatory disorders, and consider transplantation for lethal surfactant disease
chILD syndrome and its work-up — the CHILD approach
Specific Subtypes & Scenarios
The genetic surfactant dysfunction disorders are the subtype that transformed the field and are essential fellowship knowledge. Surfactant protein B deficiency and ABCA3 deficiency typically cause severe, often fatal, neonatal respiratory failure in a term infant, whereas surfactant protein C mutations more often produce chronic, variable interstitial lung disease that can appear later in infancy or childhood and run a fluctuating course. Recognising this spectrum, and testing for it early, changes prognosis, counselling, and transplant decisions. [10] [11] [12]
Neuroendocrine cell hyperplasia of infancy is the subtype to recognise for the opposite reason: it looks alarming but behaves well. It presents in a well-grown infant, usually after the first weeks of life, with persistent tachypnoea, crackles, and mild hypoxaemia, and its HRCT pattern of ground-glass change in the right middle lobe and lingula with air trapping is close to diagnostic. Recognising it avoids an unnecessary biopsy and, crucially, avoids exposing a child who will improve to the harms of immunosuppression. [5] [6]
Pulmonary interstitial glycogenosis is the other infancy-specific entity of unknown cause, often coexisting with lung growth abnormalities and presenting in the newborn period. It tends to have a relatively favourable course with supportive care, and some infants are treated with corticosteroids. Alongside the developmental and growth disorders, it completes the group of distinctly paediatric conditions that a general paediatrician should be able to name and place within the classification. [2] [9]
Complications & Pitfalls
The complications that shape outcome are chronic hypoxaemia, pulmonary hypertension, and failure to thrive, and they are interlinked. Sustained low oxygen levels drive pulmonary vascular remodelling and pulmonary hypertension, which is a leading cause of death in severe chILD, while the relentless work of breathing consumes the calories a child needs to grow. Correcting hypoxaemia and supporting nutrition are therefore not optional extras but treatments that alter the disease course. [7] [1]
The commonest pitfall is diagnostic delay from failing to think of the diagnosis, and the second is treating the category rather than the disease. Reaching reflexively for corticosteroids in an infant who actually has neuroendocrine cell hyperplasia of infancy exposes a child destined to improve to real harm, while missing a surfactant protein B deficiency in a deteriorating newborn loses the window for honest prognostication and transplant discussion. The corrective habit is precise diagnosis before directed treatment. [5] [11]
Prognosis & Disposition
Prognosis in chILD is as heterogeneous as the disorders themselves, and the single best predictor is the specific diagnosis. At one extreme, surfactant protein B and ABCA3 deficiencies are usually lethal in infancy without lung transplantation, and developmental disorders such as alveolar capillary dysplasia carry a similarly grave outlook. At the other, neuroendocrine cell hyperplasia of infancy and pulmonary interstitial glycogenosis generally improve over years with supportive care alone. [11] [5]
Between these poles sit the many chronic disorders whose course depends on the degree of hypoxaemia, the development of pulmonary hypertension, growth, and the response to any disease-specific treatment. Because the outlook and the treatment both hinge on the precise diagnosis, reaching that diagnosis through imaging, genetics, and selective biopsy is the most important prognostic act. Longitudinal follow-up then tracks oxygenation, growth, and lung function over time. [7] [8]
Disposition centres on placing every child in structured, multidisciplinary follow-up at a specialist chILD centre rather than in serial acute presentations. Each child needs a plan covering oxygen, nutrition, immunisation, monitoring for pulmonary hypertension, and genetic counselling for the family where an inherited disorder has been found. For families facing a lethal disorder, early and honest discussion of prognosis, transplantation, and palliative options is an integral part of care. [1] [4]
Special Populations
Ex-preterm infants and children with chronic neonatal lung disease sit at the overlap between bronchopulmonary dysplasia and the growth abnormalities of chILD, and the two can be hard to separate. When an ex-preterm infant has diffuse lung disease that is more severe or more persistent than the prematurity alone explains, the growth-abnormality group of chILD should be considered and the child assessed accordingly. Their care combines the principles of chronic neonatal lung disease with chILD-specific evaluation. [2] [9]
Children with immunodeficiency, whether primary or acquired through treatment and transplantation, are prone to interstitial lung disease from both infection and immune dysregulation. In this group the diffuse lung change may reflect opportunistic infection, drug toxicity, graft-versus-host disease, or an immune-mediated pneumonitis, and the work-up must weigh these possibilities carefully. Close collaboration between respiratory, immunology, and transplant teams is essential. [8] [3]
Families affected by inherited surfactant disorders have particular needs that extend beyond the index child. Because these conditions are genetic, a diagnosis carries implications for siblings and future pregnancies, so genetic counselling and, where appropriate, testing of relatives are part of comprehensive care. Delivering this alongside the emotional support required when a disorder is severe or lethal is a core responsibility of the specialist team. [10] [11]
Evidence, Guidelines & Regional Differences
The modern approach to chILD rests on two landmark developments: an agreed classification and a formal clinical guideline. The classification scheme applied by the chILD Research Cooperative to diffuse lung disease in young children gave the field a shared language organised around age and mechanism, and it remains the framework used to categorise these disorders. It is the evidence base for the age-split classification that structures this topic. [2] [9]
The American Thoracic Society clinical practice guideline on chILD in infancy then codified the chILD syndrome, the exclusion of common causes, and the sequence of HRCT, genetics, and biopsy that defines contemporary evaluation. In Europe, the chILD-EU collaboration produced consensus protocols for diagnosis and initial treatment and built the register and research infrastructure that continues to advance the field. Together these documents anchor practice on both sides of the world. [1] [4]
The genuine controversies reflect how young and rare this field is. Because randomised trials are almost impossible, the use of corticosteroids, hydroxychloroquine, and azithromycin rests on observational data, and the optimal regimens remain uncertain. The role and timing of lung biopsy continue to narrow as genetic testing improves, and the boundaries of some entities are still being refined as registers accumulate cases. The direction of travel is toward more genetic diagnosis and less invasive evaluation. [7] [8]
Exam Pearls
The highest-yield spine to recite is the pathway. A young child with persistent tachypnoea, diffuse crackles, hypoxaemia, and failure to thrive who does not fit the common diagnoses has the chILD syndrome, and the response is to exclude the common mimics, then characterise with HRCT, genetics, and selective biopsy. Holding that sequence lets you answer almost any chILD question by walking the examiner through it. [1] [4]
The second set-piece is the age-based classification and its two paradigm diseases. Learn the infancy-predominant group — developmental, growth, surfactant, and NEHI/PIG disorders — and contrast the lethal genetic surfactant deficiencies against the benign, self-improving neuroendocrine cell hyperplasia of infancy. That single contrast captures the range of prognosis and the principle that treatment must be matched to the precise diagnosis rather than to the chILD label. [2] [5]
Finally, show the management instincts an examiner rewards. Supportive care with oxygen and nutrition changes outcomes for every child; disease-specific corticosteroids and hydroxychloroquine are reserved for inflammatory disorders; NEHI needs neither; and lethal surfactant disease needs early transplant consideration and honest family counselling. Demonstrating that you would diagnose precisely, treat proportionately, and refer to a specialist chILD centre is what marks the competent candidate. [7] [11]
References
- [1]Kurland G, Deterding RR, Hagood JS, et al An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med, 2013.PMID 23905526
- [2]Deutsch GH, Young LR, Deterding RR, et al Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med, 2007.PMID 17885266
- [3]Clement A, Eber E Interstitial lung diseases in infants and children. Eur Respir J, 2008.PMID 18310399
- [4]Bush A, Cunningham S, de Blic J, et al European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax, 2015.PMID 26135832
- [5]Deterding RR, Pye C, Fan LL, et al Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol, 2005.PMID 15965897
- [6]Young LR, Brody AS, Inge TH, et al Neuroendocrine cell distribution and frequency distinguish neuroendocrine cell hyperplasia of infancy from other pulmonary disorders. Chest, 2011.PMID 20884725
- [7]Griese M Chronic interstitial lung disease in children. Eur Respir Rev, 2018.PMID 29436403
- [8]Nathan N, Berdah L, Delestrain C, et al Interstitial lung diseases in children. Presse Med, 2020.PMID 32563946
- [9]Fan LL, Deterding RR, Langston C Pediatric interstitial lung disease revisited. Pediatr Pulmonol, 2004.PMID 15376335
- [10]Nogee LM, Dunbar AE 3rd, Wert SE, et al A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med, 2001.PMID 11207353
- [11]Shulenin S, Nogee LM, Annilo T, et al ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med, 2004.PMID 15044640
- [12]Nogee LM, de Mello DE, Dehner LP, et al Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med, 1993.PMID 8421459