Paeds · respiratory-sleep-and-airway
Respiratory manifestations of systemic disease
Also known as Lung involvement in systemic disease in children · Secondary lung disease in children · Pulmonary manifestations of systemic illness · Sickle cell acute chest syndrome · Connective tissue disease-associated lung disease
Fellowship guide to the lung as a target organ of systemic disease in children — the sickle cell chest that can kill in hours, the connective tissue diseases that scar the interstitium, the immunodeficiencies that let infection and bronchiectasis take hold, and the malignancies and their treatments that infiltrate, compress, and injure the growing lung.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A ten-year-old with sickle cell disease comes in with a fever and chest pain, and the intern books him for a routine chest infection review in the morning. That decision can be fatal, because the same child can develop acute chest syndrome overnight and die of it. The single most important habit in this topic is to stop treating the lung as an innocent bystander and start treating it as the organ where a child's systemic disease often declares itself most dangerously. [1] [2]
Respiratory manifestations of systemic disease are the ways a disease rooted outside the lung — in the blood, the immune system, the connective tissues, or a malignancy — damages the airways, the alveoli, the interstitium, the pleura, or the pulmonary vessels. The chest problem is a symptom of the systemic disease, so it is understood and treated by working back to the disease that caused it rather than as an isolated pneumonia or wheeze. [7] [6]
The reason this matters for the exam and the ward is that these children fall between specialties. The haematologist, rheumatologist, immunologist, and oncologist each own the systemic disease, but the child in front of you is short of breath, and the paediatrician has to recognise the lung complication and start the right pathway. Missing the link is how a treatable sickle chest, an evolving interstitial lung disease, or an opportunistic pneumonia gets managed as a simple infection until it is too late. [2] [12]
Framed this way, the topic becomes one clinical reflex applied across four disease groups. Whenever a child with a known systemic illness develops new breathlessness, hypoxia, chest pain, or a persistent infiltrate, ask what that particular disease does to the lung, and investigate for it deliberately. The same reflex catches the child in whom the lung complication is the very first sign of an undiagnosed systemic disease. [11] [7]
Classification
The useful way to hold this topic is by the system that is failing, because the culprit disease predicts the pattern of lung injury and the treatment. Four groups account for the overwhelming majority of paediatric practice: haematological disease, connective tissue and autoimmune disease, immunodeficiency, and malignancy with its treatments. Naming the group is the first diagnostic move, because it tells you what to look for and who to involve. [7] [2]
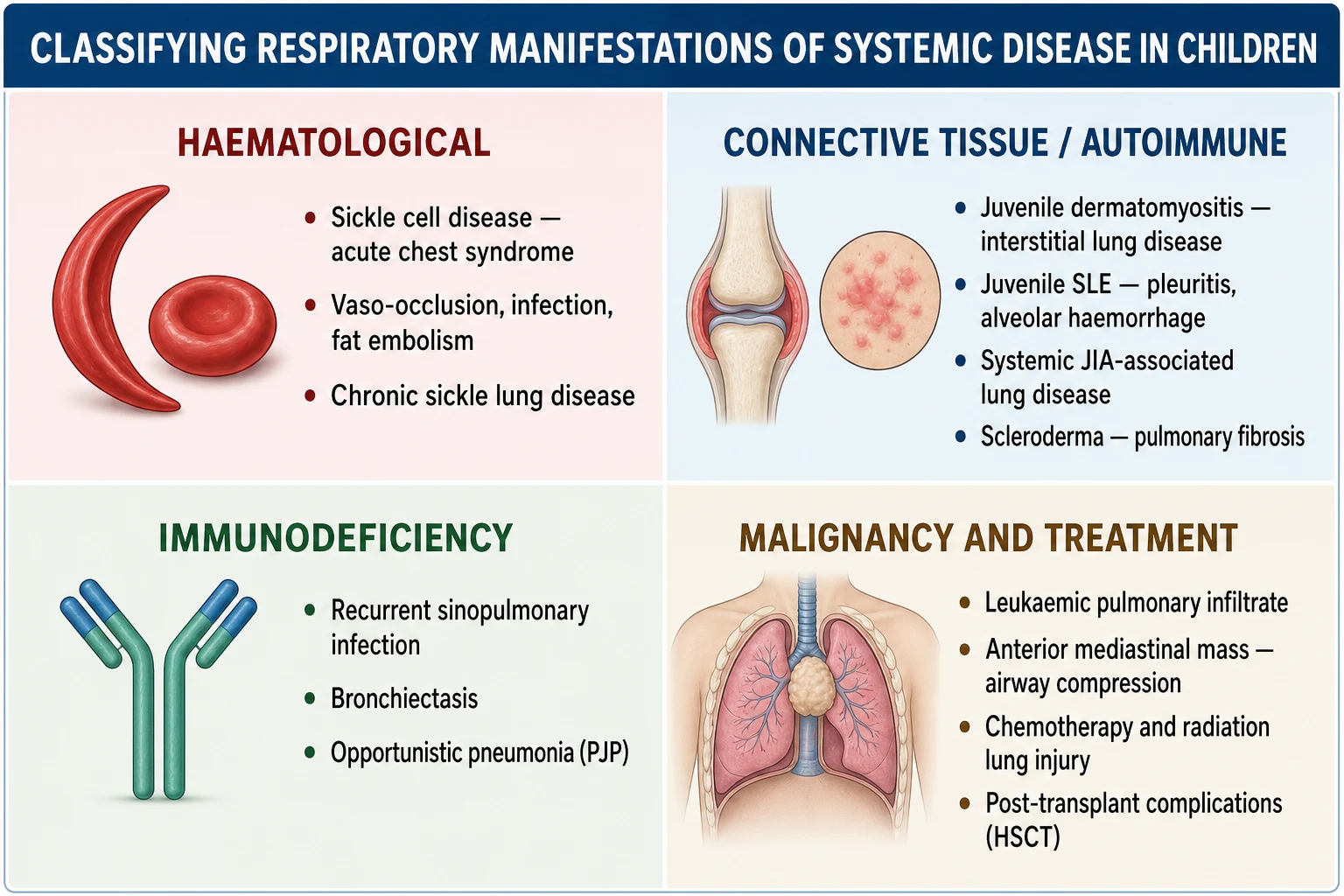
The haematological group is dominated by sickle cell disease and its acute chest syndrome, alongside chronic sickle lung disease and pulmonary hypertension. The connective tissue and autoimmune group scars and inflames the lung, producing interstitial lung disease, pleuritis and effusions, diffuse alveolar haemorrhage, and sometimes pulmonary hypertension. Each pattern points back toward a specific rheumatic diagnosis such as juvenile dermatomyositis, lupus, scleroderma, or systemic juvenile idiopathic arthritis. [8] [10]
The immunodeficiency group lets infection gain a foothold, so it presents as recurrent, severe, or unusual sinopulmonary infection and, over time, bronchiectasis. The malignancy group is broader, spanning direct leukaemic or lymphomatous infiltration, an airway-compressing mediastinal mass, opportunistic infection during immunosuppression, and lung injury from chemotherapy, radiation, and haematopoietic stem cell transplantation. Sorting a child into the right group turns a bewildering differential into a focused plan. [11] [13]
Epidemiology & Risk Factors
These complications are common precisely because the underlying diseases are, and because the lung is so often in the firing line. In sickle cell disease acute chest syndrome affects the majority of children at some point, is the commonest reason for intensive care, and is the leading cause of death, which is why every febrile or chest-symptomatic sickle child is watched for it. The risk rises with younger age, asthma, recent surgery, and a preceding painful vaso-occlusive crisis. [1] [2]
The connective tissue diseases carry disease-specific lung risk. Interstitial lung disease is a recognised and serious complication of juvenile dermatomyositis, particularly with anti-MDA5 antibodies, and of scleroderma and mixed connective tissue disease, while lupus more often causes pleuritis and, less commonly, alveolar haemorrhage. A newly described systemic juvenile idiopathic arthritis-associated lung disease has emerged, linked to macrophage activation and to biologic therapy, and it can be severe. [8] [10]
The immunodeficiency and malignancy groups share a common thread of infection risk. In inborn errors of immunity, recurrent sinopulmonary infection and bronchiectasis are frequent, and bronchiectasis in this setting predicts worse cardiopulmonary outcomes. In childhood cancer, the risks come from the disease burden, from chemotherapy and radiation, and above all from the profound immunosuppression of treatment and of haematopoietic stem cell transplantation, which opens the door to opportunistic and non-infectious lung complications. [11] [12]
Pathophysiology
Acute chest syndrome is the mechanism to be able to draw, because it is a self-amplifying loop that turns a manageable child into a critical one within hours. It begins with any trigger that lowers oxygen or splints the chest — infection, a fat embolus from infarcting marrow, or the hypoventilation of rib and sternal pain. Low oxygen promotes sickling in the pulmonary vessels, sickling causes vaso-occlusion and more infarction, and the resulting inflammation and atelectasis worsen oxygenation, which drives yet more sickling. [1] [2]
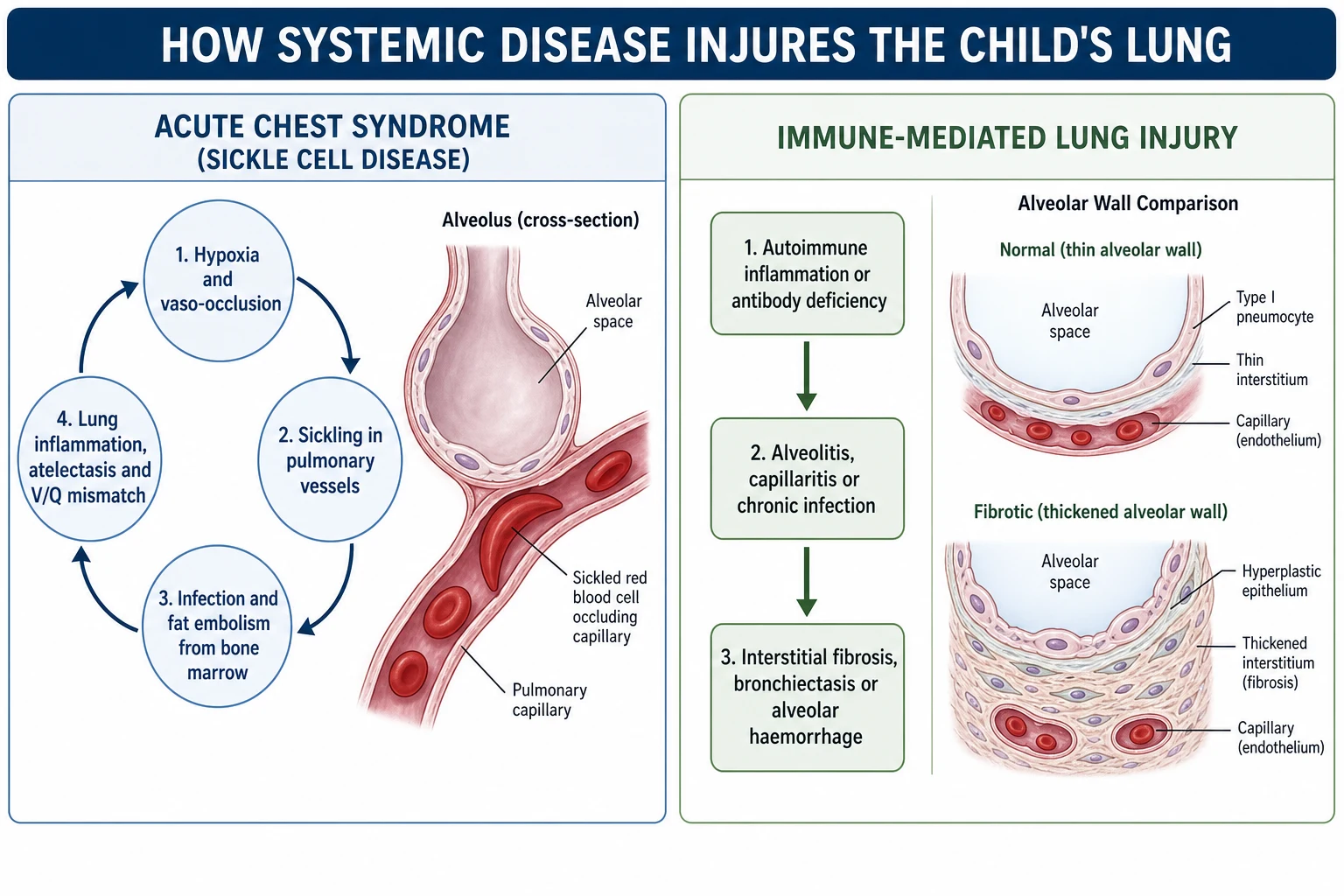
The connective tissue diseases injure the lung by immune-mediated inflammation of its tissues. Autoantibodies and activated immune cells attack the alveolar walls, the small vessels, or the pleura, producing alveolitis, capillaritis, or serositis. When alveolar inflammation goes unchecked it lays down collagen, so the thin gas-exchanging interstitium thickens into fibrosis, and once the architecture is scarred the loss of function is permanent. This is why early recognition and suppression of the inflammation matter so much. [8] [7]
Immunodeficiency and malignancy both converge on a lung that cannot defend itself. In an inborn error of immunity, absent antibody or defective cellular immunity allows ordinary and opportunistic organisms to establish chronic infection, and the same vicious cycle of infection, inflammation, and airway damage that underlies any bronchiectasis takes hold. In cancer, malignant cells can infiltrate the lung directly or compress the airway as a mass, while chemotherapy, radiation, and transplantation add direct tissue toxicity and deep immunosuppression on top. [11] [13]
Clinical Presentation
The presentation that must never be underestimated is the febrile or chest-symptomatic child with sickle cell disease. Acute chest syndrome is defined by a new pulmonary infiltrate on chest imaging together with fever or respiratory signs such as chest pain, cough, tachypnoea, wheeze, or hypoxia. Crucially, it often evolves after admission for a painful crisis, so a child who arrived with limb or back pain and then develops chest signs is declaring acute chest syndrome, not catching a cold. [1] [2]
The connective tissue diseases tend to present more insidiously in the lung. Progressive exertional breathlessness, a persistent dry cough, reduced exercise tolerance, and digital clubbing suggest evolving interstitial lung disease, whereas pleuritic chest pain and effusions point to serositis, and sudden breathlessness with anaemia and haemoptysis suggests diffuse alveolar haemorrhage. Because the lung change can precede or outpace the joint or skin disease, these respiratory symptoms deserve attention in their own right. [8] [9]
The immunodeficiency and malignancy groups announce themselves through infection and mass effect. Recurrent, severe, or unusual sinopulmonary infection, poor recovery between illnesses, and the wet cough of early bronchiectasis point toward an immune defect. In malignancy, look for the persistent infiltrate that does not resolve on antibiotics, and for the red-flag combination of a mediastinal mass with orthopnoea, stridor, facial swelling, or venous distension that signals airway and great-vessel compression. [11] [14]
Differential Diagnosis
The first question is always whether the chest problem belongs to the systemic disease or is a simple, unrelated illness. In a sickle cell child with fever and an infiltrate, the differential of acute chest syndrome overlaps almost completely with pneumonia and pulmonary infarction, and because they coexist and are managed together, the safe move is to treat for acute chest syndrome rather than to try to separate them. Over-calling it is far safer than missing it. [2] [1]
In a child with a rheumatic disease and new respiratory signs, the differential is infection versus disease activity versus drug toxicity, and the three can look identical. An immunosuppressed child with breathlessness and an infiltrate could have opportunistic infection, a flare of interstitial lung disease, or lung injury from methotrexate or another agent, and distinguishing them usually requires imaging, lung function, and often bronchoscopy rather than clinical judgement alone. [7] [10]
The trap to avoid is anchoring on the commonest, most comfortable label — a viral chest infection or asthma — in a child whose systemic disease makes a specific complication far more likely. A persistent infiltrate that does not clear, breathlessness out of proportion to a supposed cold, or infection that is too frequent or too strange should all pull you off the easy diagnosis and toward the disease-specific work-up. [11] [13]
Clinical & Bedside Assessment
The history has two jobs: characterise the respiratory problem, and connect it firmly to the systemic disease. Establish the tempo and nature of the breathlessness, cough, and chest pain, then take a focused disease history covering the sickle phenotype and past crises, the rheumatic diagnosis and its activity, the pattern and severity of past infections, and the exact chemotherapy, radiation, and transplant history. A drug history is essential, because several agents injure the lung directly. [1] [12]
The examination is a deliberate search for both the respiratory signs and the systemic fingerprints. Measure oxygen saturation and work of breathing first, listen for focal or diffuse crackles, a pleural rub, or reduced breath sounds, and look for clubbing that suggests chronic interstitial or suppurative disease. Then look beyond the chest for the rash and weakness of dermatomyositis, the tight skin of scleroderma, the growth failure of chronic disease, and the pallor and jaundice of haemolysis. [8] [9]
One bedside decision carries unusual weight: the child with a large anterior mediastinal mass. Before any sedation or anaesthesia, assess for orthopnoea, stridor, and signs of superior vena cava obstruction, because these predict catastrophic airway or cardiovascular collapse when the child lies flat or is paralysed. This single assessment can be the difference between a safe diagnostic pathway and a peri-procedural arrest. [14]
Investigations
Investigation follows the same logic as assessment: confirm and characterise the lung problem, then pin it to the systemic disease. The chest radiograph is the first-line test and defines the infiltrate of acute chest syndrome, an effusion, or a mediastinal mass, while pulse oximetry and blood gases quantify the gas-exchange problem. In acute chest syndrome, imaging and clinical criteria are enough to start treatment, and you never wait for a perfect diagnosis before acting. [1] [2]
For chronic and immune-mediated disease, high-resolution CT and pulmonary function testing are the workhorses. High-resolution CT characterises the interstitial pattern and its extent, pulmonary function tests reveal and track a restrictive defect with reduced diffusion, and echocardiography screens for the pulmonary hypertension that complicates sickle, scleroderma, and lupus lung disease. Serial lung function is what tells you whether treatment is working before irreversible fibrosis sets in. [7] [6]
Bronchoscopy with bronchoalveolar lavage earns its place when the diagnosis is uncertain and the answer changes management, which is often in the immunocompromised child. In children with leukaemia and pulmonary infiltrates, early bronchoscopy frequently reveals a specific and treatable diagnosis such as an opportunistic infection or alveolar haemorrhage, so it is used decisively rather than late. The immune work-up, with immunoglobulins, vaccine responses, and lymphocyte subsets, is added whenever infection is recurrent, severe, or unusual. [13] [11]
Management — Resuscitation
The acutely unwell child is stabilised first, along standard airway, breathing, and circulation lines, with the systemic disease shaping each step. Give oxygen to correct hypoxia, support breathing and escalate to non-invasive or invasive ventilation when work of breathing or gas exchange fail, and treat shock and severe anaemia. In acute chest syndrome this early aggressive support, combined with prompt disease-specific treatment, is what prevents the vicious cycle from spiralling into respiratory failure. [2] [3]
Two scenarios are genuine emergencies that override the usual order. The first is severe acute chest syndrome with worsening hypoxia, where urgent transfusion — often exchange transfusion — is needed to reduce the proportion of sickle haemoglobin and break the cycle, alongside oxygen, analgesia, antibiotics, and respiratory support. The second is diffuse alveolar haemorrhage in lupus or vasculitis, which needs airway protection, resuscitation, and urgent immunosuppression. [1] [9]
The most dangerous resuscitation trap in this topic is the child with a large anterior mediastinal mass who needs a procedure. Sedation, general anaesthesia, and lying flat can collapse a compressed airway or great vessels and cause arrest that is very hard to reverse. The safe approach secures the diagnosis by the least invasive route under local anaesthesia where possible, and involves senior anaesthetic and surgical teams before any sedation. [14]
Management — Definitive & Stepwise
Definitive management always pairs treatment of the lung with treatment of the disease that injured it, so the plans run in parallel. In acute chest syndrome the pillars are oxygen, adequate analgesia that allows deep breathing without oversedation, antibiotics covering typical and atypical organisms, judicious fluids, incentive spirometry, and transfusion for hypoxia or progression. Getting analgesia right matters, because pain-driven hypoventilation is a key driver, yet excess opioid suppresses breathing and worsens the syndrome. [2] [3]
Prevention is as important as rescue in sickle lung disease, and hydroxyurea is central to it. By raising fetal haemoglobin, hydroxyurea reduces painful crises and acute chest syndrome, and long-term data show it lowers morbidity and mortality, so it is offered to children with recurrent acute chest syndrome or frequent crises. Incentive spirometry during painful crises and surgery helps prevent the atelectasis that seeds acute chest syndrome, and a randomised trial supports its preventive use. [5] [4]
Hydroxyurea (disease-modifying prevention in sickle cell disease)
Dose
Started at about 20 mg/kg once daily and titrated upward toward a maximum tolerated dose while monitoring the blood count
The connective tissue diseases are managed by suppressing the inflammation before it scars, with corticosteroids as the backbone and a steroid-sparing immunosuppressant such as methotrexate, mycophenolate, cyclophosphamide, or a biologic chosen for the specific disease and severity. Rapidly progressive interstitial lung disease, especially anti-MDA5 juvenile dermatomyositis, needs early aggressive combination immunosuppression because it can be fatal, and treatment is guided by serial lung function and imaging within a rheumatology and respiratory partnership. [8] [10]
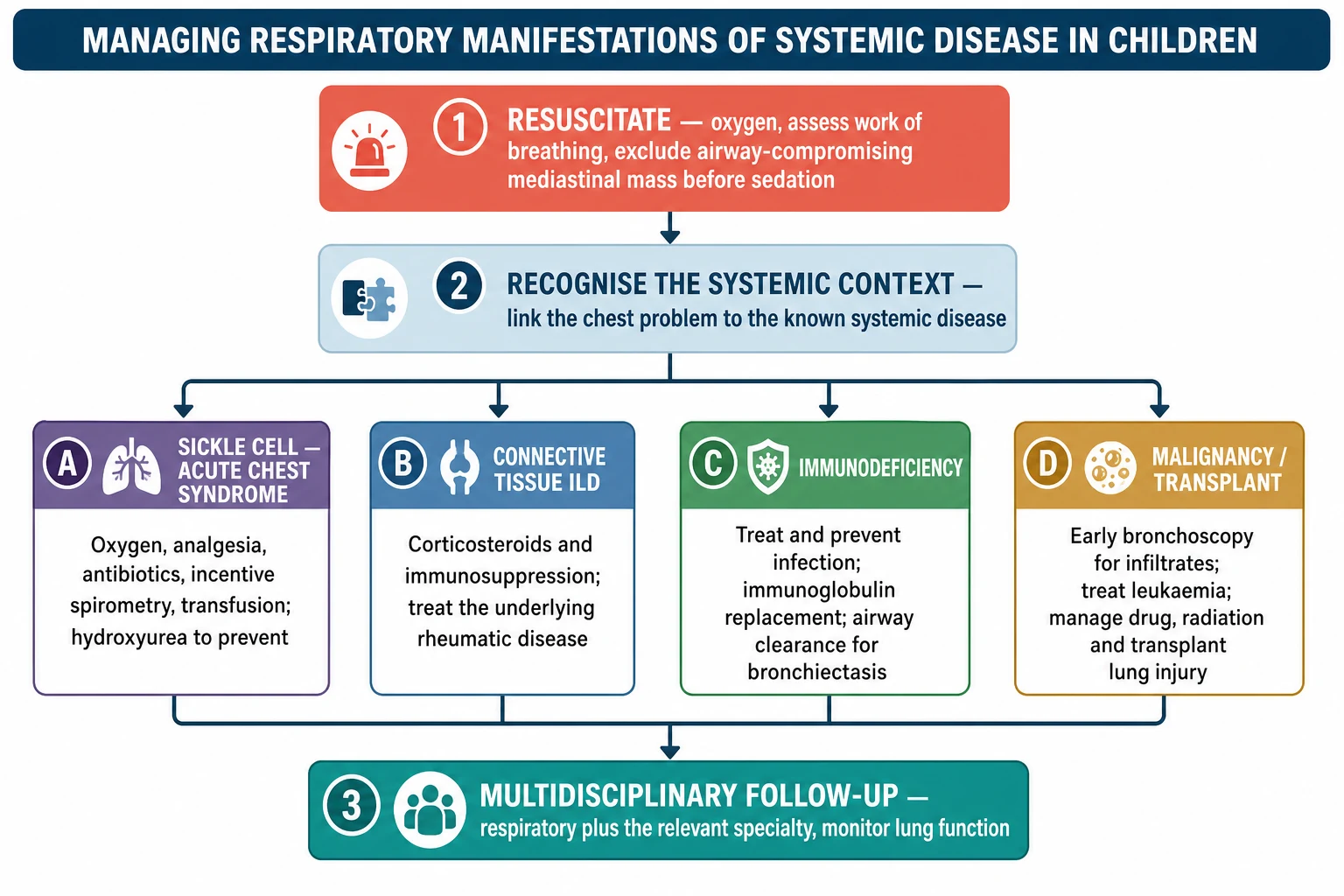
Stepwise approach to lung involvement in systemic disease
Resuscitate first and, before any sedation, exclude an airway- or vessel-compromising anterior mediastinal mass
Link the new respiratory sign to the known systemic disease rather than assuming a simple infection
In sickle cell disease treat presumptively for acute chest syndrome: oxygen, analgesia, antibiotics, incentive spirometry, and transfusion for hypoxia or progression
In connective tissue disease confirm interstitial lung disease on HRCT and lung function, then suppress inflammation early with steroids and immunosuppression
In immunodeficiency treat and prevent infection, replace immunoglobulin where indicated, and manage bronchiectasis with airway clearance
In malignancy use early bronchoscopy for infiltrates, treat the cancer, and manage drug, radiation, and transplant lung injury
Place every child in coordinated multidisciplinary follow-up with monitoring of symptoms and lung function
The immunodeficiency and malignancy groups are managed by controlling infection and treating the primary disease. Antibody deficiency is treated with immunoglobulin replacement and prompt, sometimes prophylactic, antimicrobials, and any resulting bronchiectasis is managed with airway clearance and culture-guided antibiotics. In cancer, the infiltrate is investigated early with bronchoscopy, the malignancy is treated, and drug, radiation, and transplant-related lung injuries — including bronchiolitis obliterans after transplantation — are recognised and managed with the oncology and transplant teams. [11] [12]
Groups of respiratory manifestations of systemic disease — SICK LUNGS
Specific Subtypes & Scenarios
Acute chest syndrome is the subtype that must never be missed, and its defining trap is timing. It frequently develops two to three days after admission for a painful vaso-occlusive crisis, when the initial pain has settled and vigilance drops, so any new fever, chest pain, oxygen requirement, or infiltrate in an admitted sickle child triggers the acute chest syndrome pathway immediately. The commonest identified causes in children are infection and pulmonary fat embolism, but the treatment is started before the cause is known. [1] [2]
Anti-MDA5 juvenile dermatomyositis is the connective tissue subtype that carries the gravest lung risk, because it can cause rapidly progressive interstitial lung disease with relatively subtle skin and muscle signs. A child with this antibody and even mild breathlessness deserves urgent imaging, lung function, and early aggressive immunosuppression, since delay allows fatal respiratory failure. The newly recognised systemic juvenile idiopathic arthritis-associated lung disease is a second high-stakes subtype, often severe and linked to macrophage activation. [8] [10]
The immunocompromised child with a new infiltrate is the scenario that most rewards a decisive, structured approach. The differential spans opportunistic infection, disease relapse, drug and radiation injury, and transplant-related complications, and clinical features alone rarely separate them. Early bronchoscopy with lavage in a child with leukaemia and lung infiltrates commonly yields a specific, treatable diagnosis, which is why it is used early rather than reserved for the failing patient. [13] [12]
Complications & Pitfalls
The gravest complications are death and irreversible loss of lung function. Acute chest syndrome can progress to respiratory failure and is the leading cause of death in sickle cell disease, uncontrolled interstitial inflammation scars into permanent fibrosis, and untreated immune-mediated or infective disease drives progressive bronchiectasis and pulmonary hypertension. Each of these is far more preventable than reversible, which is why the whole topic rewards early action. [1] [7]
The commonest day-to-day pitfall is diagnostic anchoring: treating each episode as an isolated infection and never stepping back to ask what the child's systemic disease does to the lung. This delays disease-specific treatment during the window when it works, and it is compounded by the peri-procedural trap of sedating a child with a compromising mediastinal mass. The corrective discipline is simple: always link the chest to the systemic disease, and always assess the mediastinum before sedation. [14] [13]
Prognosis & Disposition
Prognosis turns almost entirely on how early the lung complication is recognised and treated. Most episodes of acute chest syndrome resolve with prompt aggressive care, yet each episode injures the lung and repeated episodes contribute to chronic sickle lung disease and pulmonary hypertension, so prevention with hydroxyurea genuinely changes the long-term trajectory. Caught and treated early, the acute episode is survivable and its long-term toll is reduced. [3] [5]
In immune-mediated disease the outlook depends on catching inflammation before it becomes fibrosis. Interstitial lung disease that is treated while still inflammatory can stabilise or improve, whereas established fibrosis is permanent, and rapidly progressive forms such as anti-MDA5 disease can be fatal without early aggressive therapy. In immunodeficiency, bronchiectasis predicts worse cardiopulmonary outcomes, so preventing and controlling infection early protects long-term lung function. [8] [11]
Disposition is coordinated, multidisciplinary, long-term follow-up rather than episodic care. Each child needs the respiratory team working alongside the haematologist, rheumatologist, immunologist, or oncologist who owns the systemic disease, with a shared plan covering acute action, preventive treatment, immunisation, and regular monitoring of symptoms and lung function. Serial lung function is the instrument that detects deterioration early enough to act. [12] [7]
Special Populations
Immunocompromised and post-transplant children are the population where the stakes and the diagnostic difficulty are highest. A new infiltrate can be opportunistic infection, drug or radiation injury, disease relapse, or a transplant-related complication such as bronchiolitis obliterans, and these outcomes look alike at the bedside. Early bronchoscopy and close work with the oncology and transplant teams are what convert an undifferentiated infiltrate into a treatable diagnosis. [12] [13]
Children with complex chronic disease and technology dependence, including those with neurodisability, carry compounded respiratory risk when a systemic disease is added on top of weak cough, aspiration, or restrictive chest physiology. Their care needs an even lower threshold for imaging and specialist input, because a lung complication can decompensate limited reserve quickly. Careful attention to airway clearance and infection prevention protects that reserve. [11] [6]
Aboriginal, Torres Strait Islander, Māori, and Pacific children carry a high burden of chronic suppurative lung disease and bronchiectasis, and when an underlying immunodeficiency or systemic disease is present the risk is amplified. Equitable care means early, culturally safe recognition of persistent respiratory symptoms, a low threshold for the immune and structural work-up, and reliable access to multidisciplinary follow-up so treatable causes are not missed. [11] [2]
Evidence, Guidelines & Regional Differences
The evidence base is strongest and oldest in sickle cell disease. The National Acute Chest Syndrome Study defined the causes and outcomes of acute chest syndrome and established its central place in sickle mortality, while long-term data on hydroxyurea show reduced crises, acute chest syndrome, and mortality, and a randomised trial supports incentive spirometry for prevention. These findings anchor a management approach that is aggressive in the acute episode and preventive over the long term. [1] [5]
In childhood interstitial and immune-mediated lung disease, formal guidance and modern imaging reviews frame diagnosis and classification. An American Thoracic Society clinical practice guideline set out the classification and evaluation of childhood interstitial lung disease, and contemporary reviews map the imaging spectrum beyond infancy, while rheumatology guidance such as the juvenile lupus recommendations structures the management of the underlying disease. The emerging literature on systemic juvenile idiopathic arthritis-associated lung disease is actively reshaping practice. [6] [9]
The genuine controversies sit at the edges of newer disease. The optimal immunosuppressive regimen and its timing in rapidly progressive juvenile dermatomyositis interstitial lung disease remain debated, the recognition and treatment of systemic juvenile idiopathic arthritis-associated lung disease is still evolving, and the balance of empirical treatment versus early bronchoscopy in the immunocompromised child varies by centre and local expertise. Across all of them the direction of travel is toward earlier recognition and earlier action. [10] [12]
Exam Pearls
The highest-yield spine to recite is the core reflex and the four groups. In a child with a systemic disease, new breathlessness, hypoxia, or a persistent infiltrate is a lung complication of that disease, and you sort it into one of four groups — sickle cell, connective tissue, immunodeficiency, or malignancy and its treatment. Naming the group tells you the pattern, the investigation, and the team to call. Hold that structure and the topic organises itself. [2] [7]
The second set-piece is the two emergencies that examiners love. The first is acute chest syndrome in the sickle child, treated presumptively and early with oxygen, analgesia, antibiotics, incentive spirometry, and transfusion, and prevented long-term with hydroxyurea. The second is the anterior mediastinal mass, where sedation can be fatal, so you assess airway and cardiovascular reserve and biopsy by the safest route before any anaesthesia. [1] [14]
Finally, show the instinct that marks a safe candidate: act before the disease declares itself. Treat the sickle chest before the picture completes, image and suppress interstitial lung disease before it fibroses, work up recurrent infection before bronchiectasis is fixed, and reach for early bronchoscopy in the immunocompromised child whose infiltrate will not clear. Early recognition and coordinated multidisciplinary care are what change outcomes across every group. [12] [11]
References
- [1]Vichinsky EP, Neumayr LD, Earles AN, et al Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med, 2000.PMID 10861320
- [2]Ramirez V, Mercier-Ross J Acute Chest Syndrome in Children with Sickle Cell Disease: A Narrative Review. Children (Basel), 2026.PMID 42194196
- [3]Rice RR, Willen SM Supportive care, prevention, and emerging therapies for acute chest syndrome in sickle cell disease. Expert Rev Hematol, 2026.PMID 42446872
- [4]van Tuijn CFJ, Gaartman AE, Nur E, et al Incentive spirometry to prevent acute chest syndrome in adults with sickle cell disease; a randomized controlled trial. Am J Hematol, 2020.PMID 32242978
- [5]Steinberg MH, Barton F, Castro O, et al Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA, 2003.PMID 12672732
- [6]Kurland G, Deterding RR, Hagood JS, et al An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med, 2013.PMID 23905526
- [7]Ikeda O, Tsujioka Y, Nishimura G, et al Imaging Spectrum of Childhood Interstitial Lung Diseases: Focus on Disorders Not Specific to Infancy. Radiographics, 2026.PMID 42241322
- [8]Lepage M, Pereira G, Akhalwaya S, et al MDA5-associated juvenile dermatomyositis and interstitial lung disease from rapidly progressive to silent: a report of three cases in South African children and a review of the literature. Clin Rheumatol, 2026.PMID 42018270
- [9]Mönkemöller K, Weber LT, Häusler M, et al Interdisciplinary Clinical Practice Guidelines for patient-centred management of juvenile-onset systemic lupus erythematosus. EULAR Rheumatol Open, 2025.PMID 42367668
- [10]Unal D, Cam V, Konte EK, et al Systemic JIA-Associated Lung Disease: A Multicenter Analysis of Clinical Features, Treatment Challenges, and Outcomes. Pediatr Pulmonol, 2026.PMID 42261212
- [11]Marangu-Boore D, Myint-Hpu K, Kang E, et al Bronchiectasis in Inborn Errors of Immunity: Prevalence, Predictors, and Cardiopulmonary Complications in a Genetically Characterized Cohort. J Clin Immunol, 2026.PMID 42414785
- [12]Srikanthan MA, Cheng PC, Goldfarb SB Pulmonary complications in pediatric hematopoietic stem cell transplantation: an overview for pediatricians. Curr Opin Pediatr, 2026.PMID 41983728
- [13]Furuya ME, Ramírez-Figueroa JL, Vargas MH, et al Diagnoses unveiled by early bronchoscopy in children with leukemia and pulmonary infiltrates. J Pediatr Hematol Oncol, 2012.PMID 22322936
- [14]Garey CL, Laituri CA, Valusek PA, et al Management of anterior mediastinal masses in children. Eur J Pediatr Surg, 2011.PMID 21751123