Paeds · haematology-oncology-and-transfusion
Haemophilia A and B
Also known as Haemophilia A · Factor VIII deficiency · Haemophilia B · Christmas disease · Factor IX deficiency
Fellowship guide to haemophilia A and B: the X-linked recessive coagulopathies from deficiency of factor VIII or factor IX, the factor-level severity classification (severe under 1 percent, moderate 1 to 5 percent, mild over 5 to 40 percent), the pathophysiology of the intrinsic tenase complex, the clinical picture of spontaneous haemarthroses and intracranial haemorrhage, primary prophylaxis with factor concentrate and subcutaneous emicizumab, on-demand bleed treatment and the factor recovery rules, the diagnosis and management of inhibitors with the Bethesda assay and immune tolerance induction, and the special care of neonates, carrier females and the transitioning adolescent.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the eighteen-month-old boy brought in with a swollen, warm, painful right knee that appeared overnight after he crawled, his mother telling you he has had two similar episodes and that her brother was a "bleeder" who needed injections. That boy has severe haemophilia, and the overnight swollen joint is a haemarthrosis. The family history on the mother's side is the X-linked recessive clue, and the joint bleed is the clinical signature of secondary haemostasis failing. [1]
Haemophilia is an inherited bleeding disorder caused by a deficiency of one of the coagulation factors of the intrinsic pathway that generates thrombin. Haemophilia A is deficiency of factor VIII, encoded by the F8 gene at Xq28, and accounts for about 80 to 85 percent of cases. Haemophilia B is deficiency of factor IX, encoded by the F9 gene at Xq27, also called Christmas disease after the first patient described, and accounts for 15 to 20 percent. Both are inherited X-linked recessive, so they almost exclusively affect males, while females are usually asymptomatic carriers. The two conditions are clinically indistinguishable, share the same severity scale, and are distinguished only by the specific factor assay. [2] [3]
The defining principle is that severity is determined not by the gene mutation but by how much residual factor activity the child has left. A child with severe haemophilia has a factor level under 1 percent of normal and bleeds spontaneously into joints and muscles. A child with mild haemophilia, a level over 5 to 40 percent, bleeds only after surgery or major trauma and may not be diagnosed until adulthood. This single relationship between residual factor activity and clinical phenotype is the backbone of every management decision that follows. [1] [4]
Classification
Haemophilia is classified along two axes that together define the disease: the type (A versus B, the missing factor) and the severity (the residual factor level). The type tells you which concentrate to give; the severity tells you how often the child will bleed and how aggressive prophylaxis must be. [1]
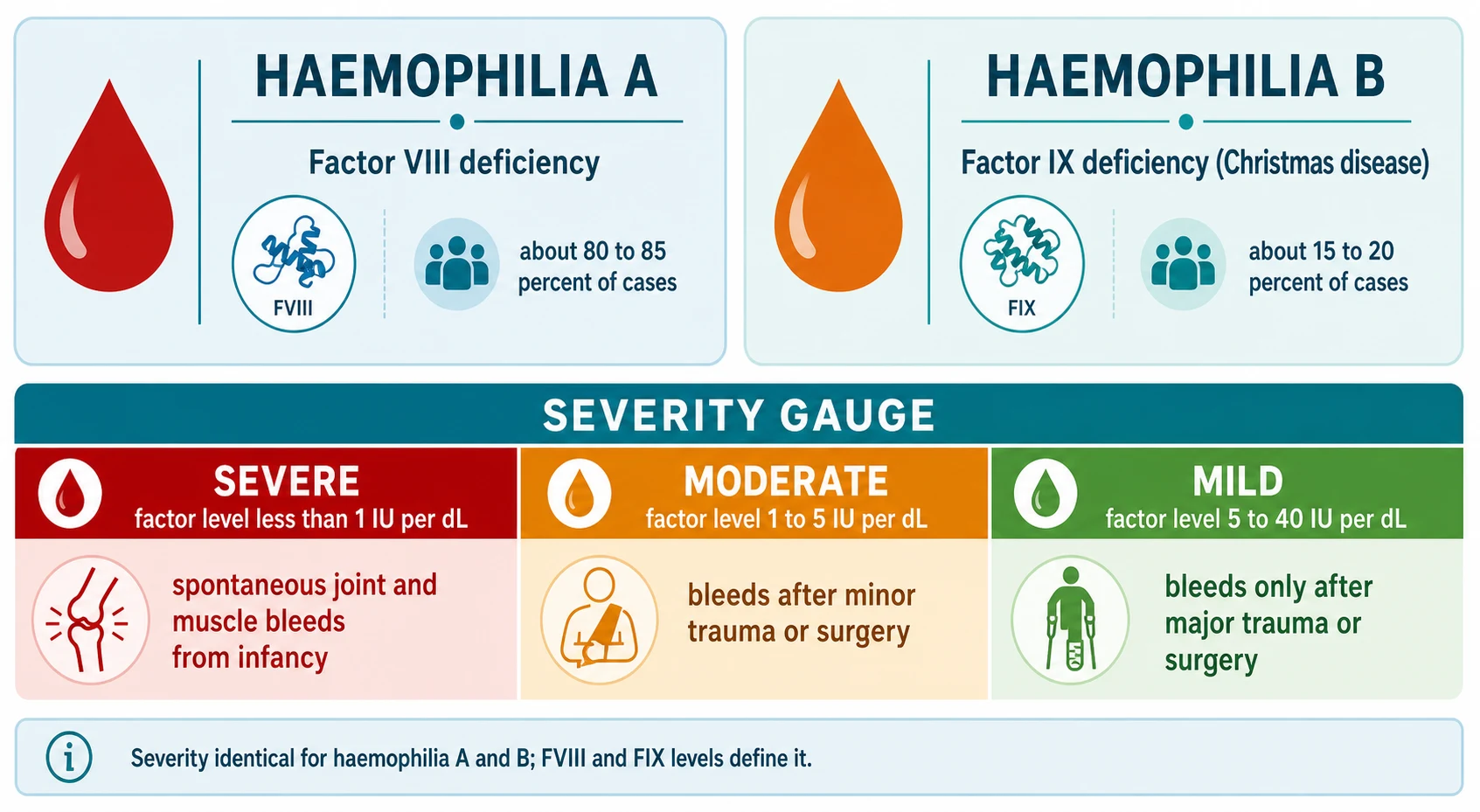
The type axis is straightforward. Haemophilia A is factor VIII deficiency and is about five times more common than haemophilia B. Both share an identical clinical phenotype, an identical severity scale, and an identical X-linked recessive inheritance. The two are separated only by the specific factor assay, which is why both factor VIII and factor IX levels are measured whenever haemophilia is suspected. [2] [3]
Severity by residual factor activity
Mild
over 5 to 40 percent
The severity axis is the one that matters clinically, and it is expressed in international units per millilitre (IU per mL) or as a percentage of normal. Severe haemophilia is a factor level under 0.01 IU per mL, that is under 1 percent of normal. Moderate haemophilia is 0.01 to 0.05 IU per mL, or 1 to 5 percent. Mild haemophilia is greater than 0.05 to 0.40 IU per mL, or over 5 to 40 percent. A normal factor level is 0.50 to 1.50 IU per mL (50 to 150 percent). The same thresholds apply to both haemophilia A and haemophilia B, and they predict the bleeding pattern with remarkable accuracy. [1]
A second, equally important classification is the inhibitor status, because the presence of a neutralising antibody to infused factor transforms the disease from a manageable condition into one of the hardest problems in paediatric haematology. About 30 percent of children with severe haemophilia A and about 3 percent of those with severe haemophilia B develop an inhibitor, usually within the first 50 exposure days. Inhibitor status is therefore a mandatory part of every haemophilia classification, and it is re-checked at every clinic visit and after each new exposure. [7] [8]
Epidemiology & Risk Factors
Haemophilia is the second commonest inherited bleeding disorder after von Willebrand disease, but it is the commonest severe inherited coagulopathy. Haemophilia A has a prevalence of about 1 in 5000 to 1 in 10000 male births, and haemophilia B about 1 in 30000 to 1 in 40000 male births. The prevalence is similar across all ethnic groups, reflecting the absence of any selective advantage or disadvantage, and the worldwide prevalence of severe haemophilia is about 1 in 16000 males. [3] [4]
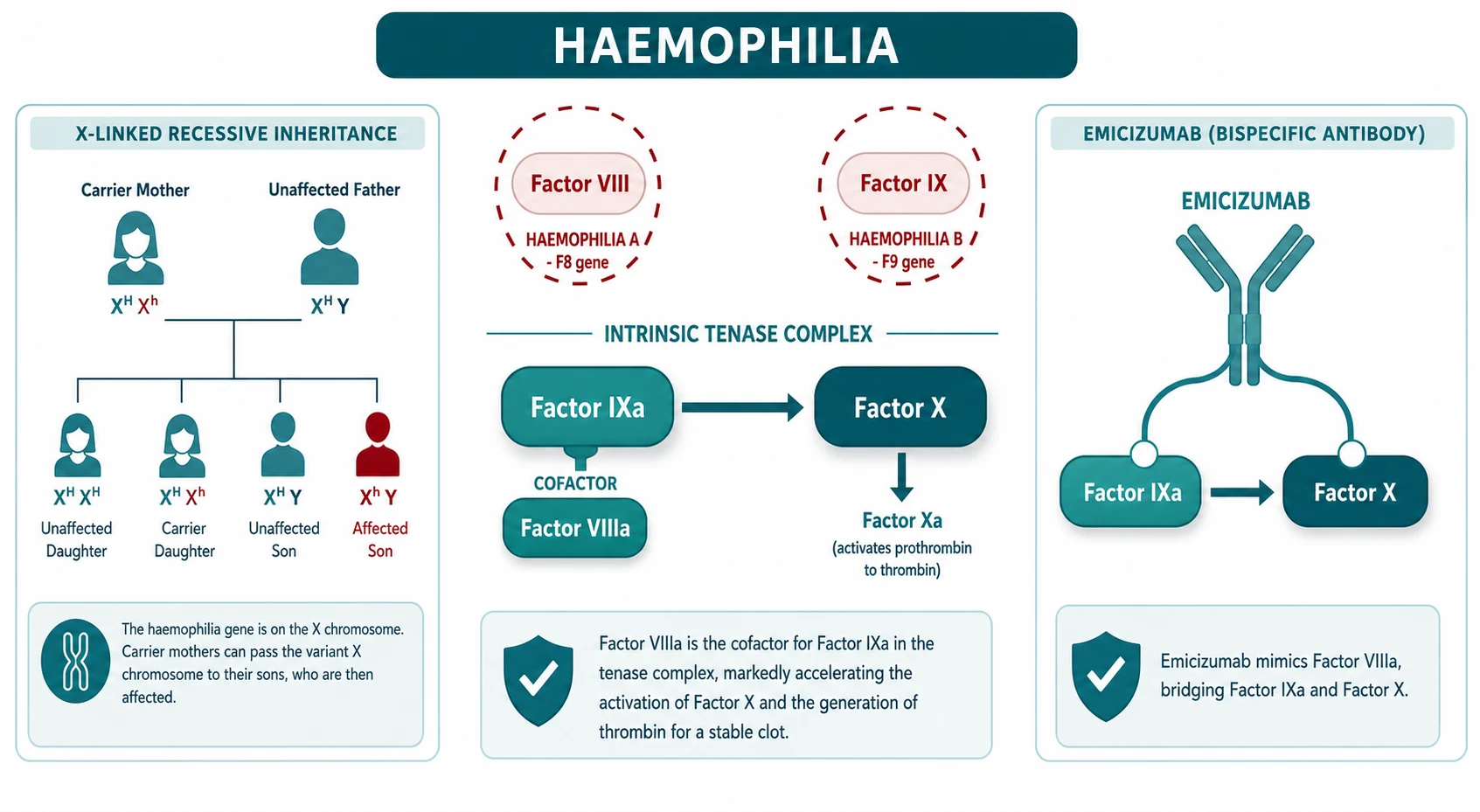
The inheritance is X-linked recessive. A carrier mother has one affected X chromosome carrying the F8 or F9 mutation and one normal X. Each son has a 50 percent chance of inheriting the affected X and having haemophilia, and each daughter has a 50 percent chance of inheriting it and being a carrier. Males cannot be asymptomatic carriers: a male with the mutation on his single X chromosome has the disease. About two-thirds of cases have a positive family history, and about one-third of severe cases arise from a de novo mutation with no family history, which is why a negative family history never excludes haemophilia. [2] [3]
The most important risk factors for a poor outcome are a delayed diagnosis, the development of an inhibitor, and inadequate prophylaxis. A delayed diagnosis is most dangerous in the neonate, where an intracranial haemorrhage after instrumental delivery can be fatal or cause permanent neurological injury, and in the child with mild disease whose first presentation is a major surgical bleed. Inhibitor development, the formation of an antibody that neutralises infused factor, is the single event that most changes the prognosis and the management. [7] [3]
Pathophysiology
Understanding haemophilia means understanding one short stretch of the coagulation cascade: the intrinsic tenase complex. Factor IX, once activated to factor IXa, sits on an activated phospholipid surface and cleaves factor X to factor Xa. Factor Xa then drives the prothrombinase complex that generates thrombin, and thrombin converts fibrinogen to the fibrin clot. This step, the activation of factor X by factor IXa, is the rate-limiting amplification step of thrombin generation. [2]
Factor IXa alone is slow. It is accelerated by several orders of magnitude by its cofactor, factor VIIIa, which holds factor IXa and factor X in the right geometry on the activated phospholipid surface. So the tenase complex is factor IXa plus factor VIIIa working together. If factor VIII is deficient, as in haemophilia A, the tenase complex assembles poorly and factor X activation collapses. If factor IX is deficient, as in haemophilia B, the same thing happens from the other side. Either way, the downstream thrombin burst is blunted, the fibrin clot is weak and delayed, and secondary haemostasis fails. [2] [4]
This explains the characteristic bleeding pattern of haemophilia. Primary haemostasis — the formation of the initial platelet plug — is intact, so the platelet count and the bleeding time are normal, and the child does not have petechiae, easy bruising or immediate mucosal oozing. What fails is secondary haemostasis, the stabilisation of the plug with fibrin. So the bleeds are delayed and deep: into joints (haemarthroses), into muscles (haematomas), and into the brain and genitourinary tract. This contrast with platelet-type bleeding (petechiae, epistaxis, mucosal ooze) is one of the most discriminating exam points. [3] [4]
The gene defects are well mapped. In haemophilia A, the F8 gene at Xq28 is large and prone to mutation. The single commonest cause of severe haemophilia A is the intron 22 inversion, an intrachromosomal recombination that accounts for about half of all severe cases; large deletions, nonsense mutations and missense mutations account for the rest. The F9 gene at Xq27 is smaller, and haemophilia B is caused by point mutations and deletions. The genotype predicts the severity and, importantly, the inhibitor risk: large F8 deletions and nonsense mutations carry the highest risk of inhibitor development. [2] [7]
[9] [10]Clinical Presentation
The clinical presentation follows the severity almost exactly, and the severity follows the factor level, so where you meet the child on the severity spectrum predicts what you will see. The severe child bleeds spontaneously and early; the mild child bleeds only after a challenge and may be discovered by chance. [1] [3]
The severe child (factor level under 1 percent) presents in infancy or early the second year of life with spontaneous bleeds. The signature bleed is the haemarthrosis, into the knees, ankles and elbows in that order, which appear as the child begins to crawl and walk. The joint becomes warm, swollen and painful, the child holds it in a position of comfort (flexion), and recurrent bleeds into the same joint cause chronic synovitis, cartilage damage and crippling haemophilic arthropathy. The other hallmark is the intramuscular haematoma, into the iliopsoas (causing a hip flexion contracture and femoral nerve compression), the forearm compartments, the calf and the buttock. [2] [4]
The neonate with severe haemophilia can present with intracranial haemorrhage after instrumental or traumatic delivery, a large cephalohaematoma, prolonged bleeding from a heel-stick or vitamin K injection, or prolonged bleeding after circumcision. Any unexplained intracranial bleed in a male neonate warrants a coagulation screen and factor assays, and any known carrier mother should have a planned delivery that avoids ventouse and forceps. [3]
The mild child (factor level over 5 to 40 percent) bleeds only after significant trauma, dental extraction or surgery. Haemophilia may not be diagnosed until an abnormal activated partial thromboplastin time turns up on a preoperative screen, or until a tonsillectomy or dental extraction bleeds far more than expected. A careful bleeding history and a low threshold for factor assays prevent these avoidable surgical bleeds. [4] [1]
| Clinical picture | What it implies | Act |
|---|
The bleeds that kill or maim are intracranial haemorrhage, airway-threatening bleeds (sublingual, retropharyngeal), and compartment-threatening limb bleeds. Intracranial haemorrhage is a leading cause of death in haemophilia and presents after even trivial head injury, so any headache, vomiting, drowsiness or seizure in a child with haemophilia is treated as an intracranial bleed first and confirmed second. [3] [12]
Differential Diagnosis
Build the differential around the single abnormal result that haemophilia produces: an isolated prolonged activated partial thromboplastin time with a normal platelet count, a normal prothrombin time and a normal fibrinogen. The mixing study corrects, proving a factor deficiency rather than an inhibitor. From there, the question is which factor is deficient and whether the cause is inherited or acquired. [3] [4]
The single most important mimic to exclude is von Willebrand disease, the commonest inherited bleeding disorder. Von Willebrand factor is the carrier protein for factor VIII in the circulation, so von Willebrand disease can produce a low factor VIII and a mildly prolonged activated partial thromboplastin time that looks like mild haemophilia A. The decisive tests are the von Willebrand factor antigen and the ristocetin cofactor activity, which are low in von Willebrand disease and normal in haemophilia A. Always measure these before labelling a child as mild haemophilia A. [3] [2]
Haemophilia A
FVIII under 40 percent
- X-linked recessive, males
- Joint and muscle bleeds
- Factor VIII low, FIX normal
- VWF antigen and activity normal
Haemophilia B
Christmas disease
- X-linked recessive, males
- Identical bleeding phenotype
- Factor IX low, FVIII normal
- Anaphylaxis risk with inhibitor
Von Willebrand disease
- Autosomal, both sexes
- Mucocutaneous bleeding
- Low VWF antigen and activity
- Factor VIII may be low
Factor XI deficiency
- Autosomal recessive
- Variable bleeding
- Post-surgical bleeding
- Normal FVIII and FIX
The other inherited factor deficiencies that prolong the activated partial thromboplastin time are factor XI deficiency (autosomal recessive, Ashkenazi Jewish predilection, variable bleeding), factor XII deficiency (which prolongs the APTT but causes no bleeding at all, a classic exam trap), and the rarer deficiencies of factors that overlap the intrinsic and common pathways. Acquired haemophilia, an autoantibody to factor VIII in older patients or postpartum women, presents with sudden severe bleeding and a mixing study that does not correct. [3]
The final layer is the artefact. Heparin contamination from a line draw prolongs the activated partial thromboplastin time and is excluded by drawing from a fresh peripheral sample. A transiently low factor VIII from a severe illness or vitamin K deficiency can mimic mild haemophilia and resolves on repeat testing. The diagnosis of haemophilia requires a persistent, reproducible factor deficiency with a normal von Willebrand screen. [4] [12]
Clinical & Bedside Assessment
Assessment begins with a structured bleeding history, because the pattern of bleeding tells you the mechanism. Use a validated bleeding assessment tool and ask specifically about joint and muscle bleeds, bleeding after circumcision or dental work, surgical bleeding, intracranial bleeds, and the matrilineal family history of affected males, carrier females or unexplained male deaths in infancy. A boy with a history of two or three swollen joints, a mother whose brother needed injections for bleeding, and a maternal uncle who died of a brain bleed has severe haemophilia until proven otherwise. [3] [1]
The musculoskeletal examination is where the long-term damage shows. Examine every major joint for swelling, warmth, synovial thickening, limited range of motion, fixed flexion deformity and the surrounding muscle wasting of chronic haemophilic arthropathy. The target joints — usually a knee, ankle or elbow that has bled repeatedly — develop a characteristic chronic synovitis. Feel the calves, forearms and hips for the firm tender swellings of intramuscular haematomas, and check for a hip flexion contracture that signals an iliopsoas bleed. [4]
[1] [3]Assess the child's overall growth, development and immunisation status, and the family's understanding of the disease. Note the central venous access — many children on prophylaxis have a portacath, and the site and patency of that line matter at every visit. Ask about school attendance, because frequent bleeds and hospital visits disrupt schooling, and screen for the anxiety and depression that are commoner in adolescents with chronic haemophilia. [4]
Investigations
The investigation strategy serves three purposes: to confirm the coagulopathy, to identify the deficient factor and its level, and to screen for inhibitors. The first test is the coagulation screen, which in haemophilia shows an isolated prolonged activated partial thromboplastin time with a normal platelet count, a normal prothrombin time and a normal fibrinogen. The mixing study, which mixes the patient's plasma with normal plasma, corrects the prolonged activated partial thromboplastin time, proving a factor deficiency rather than an inhibitor. [3] [4]
The haemophilia coagulation profile
The specific factor assays confirm the diagnosis and classify the severity. Measure both factor VIII and factor IX activity, expressed in IU per mL or as a percentage of normal, because the two conditions are clinically identical and are distinguished only by the assay. Always measure the von Willebrand factor antigen and ristocetin cofactor activity alongside the factor VIII, because von Willebrand disease is the single most important mimic of mild haemophilia A and is missed if only factor VIII is sent. [3] [1]
Factor VIII and factor IX concentrate
Dose
Factor VIII: one IU per kg raises the level by about 2 percent (0.02 IU per mL); Factor IX: one IU per kg raises the level by about 1 percent (0.01 IU per mL); So a 20 kg child given factor VIII 1000 IU (50 IU per kg) reaches about 100 percent; And a 20 kg child given factor IX 2000 IU (100 IU per kg) reaches about 100 percent
The third purpose is inhibitor screening. Every child with a new diagnosis of haemophilia is screened for an inhibitor at diagnosis and after each exposure to factor in the first 50 exposure days, and at least six-monthly thereafter. The inhibitor is measured by the Nijmegen modification of the Bethesda assay, and a significant inhibitor titre is at or above 0.6 Bethesda units. A child who bleeds more than expected or who fails to respond to the usual factor dose must have a Bethesda assay checked urgently. [7] [1]
Genetic testing identifies the F8 or F9 mutation and is useful for family counselling, for predicting inhibitor risk (large deletions and nonsense mutations carry the highest risk), and for identifying carrier females in the family. The intron 22 inversion is found in about half of severe haemophilia A families. Molecular testing of at-risk female relatives confirms carrier status and informs reproductive counselling. [2] [7]
Management — Resuscitation
The resuscitation principle in haemophilia is simple and absolute: for any serious or life-threatening bleed, give the specific factor concentrate immediately to bring the level to near 100 percent, and do not wait for confirmatory tests. An intracranial bleed can be fatal within hours, and the half-life of factor VIII is only about 12 hours, so a delay to obtain a factor level or a scan can cost the child's life or neurological future. [1] [3]
Resuscitation of a suspected intracranial or major bleed in haemophilia
Recognise: headache, drowsiness, seizure, airway swelling or compartment after trauma — assume the worst
Give factor immediately: factor VIII 50 IU per kg (to about 100 percent) or factor IX 100 IU per kg (to about 100 percent)
Do NOT wait for a factor level, a mixing study or a CT scan before the first dose
Then arrange imaging (CT brain) and admission; repeat factor at the appropriate interval to maintain the level
Alert the haemophilia treatment centre and the haematologist for ongoing management
For the child with a known inhibitor who has a major bleed, standard factor concentrate will not work, and the resuscitation agent is a bypassing agent — recombinant activated factor VII (rFVIIa) at 90 micrograms per kg every two hours, or an activated prothrombin complex concentrate such as FEIBA at 50 to 100 IU per kg. A child on emicizumab prophylaxis who has a breakthrough bleed still needs additional factor concentrate, because emicizumab maintains only a baseline haemostatic level and does not cover a major bleed on its own. [9] [12]
The general resuscitation measures apply alongside factor: airway protection for airway-threatening bleeds, analgesia, immobilisation of a bleeding limb, and avoidance of intramuscular injections and aspirin or non-steroidal anti-inflammatory drugs, which worsen bleeding. A child with haemophilia should never receive an intramuscular injection; vaccines and analgesia go subcutaneously or intravenously. [1]
Management — Definitive & Stepwise
The boy with severe haemophilia will bleed again and again unless something prevents it, and the whole of modern haemophilia care is built around stopping that next bleed. Definitive management runs along three lines: prophylaxis to prevent bleeds, on-demand treatment when a bleed happens, and the inhibitor pathway for the child who has developed an antibody. Modern prophylaxis has transformed haemophilia from a disease of crippling arthropathy and early death into a chronic condition compatible with a near-normal life, and the choice between factor concentrate and emicizumab is the central decision in current care. [1] [4]

Primary prophylaxis is regular factor replacement started early to prevent bleeds and joint damage. The landmark Joint Outcome Study (Manco-Johnson 2007) showed that primary prophylaxis with factor VIII, started before 30 months of age and before the second joint bleed, preserved joint structure on magnetic resonance imaging compared with episodic treatment, and the ESPRIT trial confirmed that prophylaxis reduces bleeding and joint damage. The World Federation of Hemophilia recommends primary prophylaxis for all children with severe haemophilia. [5] [6]
The conventional prophylaxis regimen for severe haemophilia A is factor VIII concentrate 25 to 40 IU per kg every 48 hours or three times weekly, reflecting the short half-life of factor VIII of about 12 hours. For severe haemophilia B, the longer half-life of factor IX of about 18 to 24 hours allows factor IX concentrate 40 to 100 IU per kg given twice weekly. Extended half-life factor concentrates lengthen these intervals and reduce infusion burden, which is especially valuable for children with poor venous access. [1] [4]
The Joint Outcome Study (Manco-Johnson 2007) and ESPRIT (Gringeri 2011)
Randomised trials of primary prophylaxis versus episodic factor VIII in boys with severe haemophilia A
Key finding
Primary prophylaxis started before 30 months preserved joint structure on MRI and reduced bleeding; ESPRIT confirmed fewer bleeds and less joint damage with prophylaxis.
Practice change
Primary prophylaxis is now standard of care for all children with severe haemophilia, started before the second joint bleed.
The modern alternative for haemophilia A is emicizumab, a bispecific antibody that mimics factor VIIIa by bridging activated factor IX and factor X. It is given subcutaneously with a loading dose of 3 mg per kg weekly for four weeks, followed by a maintenance dose of 1.5 mg per kg weekly, 3 mg per kg every two weeks, or 6 mg per kg every four weeks. The HAVEN trials showed that emicizumab prophylaxis dramatically reduced annualised bleed rates in patients with and without inhibitors, and the paediatric HAVEN 2 study confirmed efficacy and a subcutaneous route acceptable to young children. Emicizumab has become first-line prophylaxis for many children with haemophilia A in Australia and New Zealand. [9] [10] [11]
For breakthrough bleeds in a child not on prophylaxis, or for breakthrough bleeds despite prophylaxis, give on-demand factor concentrate at the dose the bleed requires. A minor bleed (early joint) needs factor VIII 20 IU per kg; a moderate joint or muscle bleed needs factor VIII 30 to 40 IU per kg; a major bleed (intracranial, surgical, iliopsoas) needs factor VIII 50 IU per kg to reach near 100 percent. For haemophilia B, the same clinical targets translate to higher factor IX doses because of the lower recovery: 40 to 80 IU per kg for most bleeds and 100 IU per kg for life-threatening bleeds. Apply rest, ice, compression and elevation, and review the child at 24 to 48 hours. [1] [12]
For mild haemophilia A, desmopressin (DDAVP) is first-line for minor bleeds and minor surgery. A dose of 0.3 micrograms per kg intravenously over 30 minutes releases stored factor VIII and von Willebrand factor from endothelial cells, typically raising the factor VIII level by two to three times, and avoids the need for factor concentrate. Tranexamic acid, at 15 mg per kg orally or 10 mg per kg intravenously three times daily, is a useful adjunct for mucosal and dental bleeding and is combined with factor for oral surgery. [4] [1]
Specific Subtypes & Scenarios
Severe haemophilia presenting in infancy is the commonest scenario and sets the pattern for life. The first spontaneous haemarthrosis usually appears between nine and eighteen months as the child becomes mobile, and the priority is to start primary prophylaxis early, before the second joint bleed, to prevent target-joint formation and arthropathy. Most infants need a central venous access device (a portacath) for regular factor infusion, because peripheral venous access in a toddler is difficult and traumatic; emicizumab, being subcutaneous, avoids this need and is now preferred where available. [5] [11]
Mild haemophilia discovered at surgery or after trauma is a frequent first presentation and a test of preoperative assessment. Any child with an unexplained prolonged activated partial thromboplastin time before surgery must have factor assays and a von Willebrand screen sent and the result reviewed before the operation. For a surgical procedure in mild haemophilia A, desmopressin plus tranexamic acid may suffice for minor surgery, while major surgery needs factor VIII concentrate to maintain a level near 100 percent for the perioperative period and a falling trough thereafter. [4] [1]
Haemophilia with inhibitors is the hardest subtype to manage. The inhibitor is a neutralising antibody that destroys infused factor, so standard factor concentrate stops working and the child bleeds uncontrollably. Acute bleeds are treated with bypassing agents — recombinant activated factor VII at 90 micrograms per kg every two hours, or activated prothrombin complex concentrate (FEIBA) at 50 to 100 IU per kg — which generate thrombin downstream of the blocked step. Long-term, the inhibitor is managed with emicizumab prophylaxis to prevent bleeds and, in many centres, with immune tolerance induction to eradicate the antibody. [9] [12]
JOINT
Complications & Pitfalls
The complications of haemophilia divide into those of the disease and those of its treatment. The dominant disease complication is haemophilic arthropathy, the progressive joint destruction caused by recurrent haemarthroses that deposit iron in the synovium and cartilage. Arthropathy is the leading cause of long-term morbidity, and preventing it is the entire purpose of primary prophylaxis. The other major disease complications are intracranial haemorrhage (a leading cause of death), compartment syndrome from muscle bleeds, and airway-threatening bleeds into the neck and floor of the mouth. [3] [4]
The treatment complications begin with the inhibitor, which transforms a manageable disease into one of the most challenging in paediatric haematology. Inhibitors develop in about 30 percent of children with severe haemophilia A and about 3 percent of those with severe haemophilia B, usually within the first 50 exposure days. In haemophilia B inhibitors, exposure to factor IX can trigger severe anaphylactoid reactions, and immune tolerance induction can cause nephrotic syndrome, which makes haemophilia B inhibitors especially dangerous. [7] [8]
The complications of emicizumab are the new chapter. When emicizumab is combined with activated prothrombin complex concentrate above 50 IU per kg per day for more than 24 hours, there is a risk of thrombotic microangiopathy, so bypassing agent doses must be limited and monitored. Emicizumab also shortens the activated partial thromboplastin time artefactually, so standard APTT-based factor monitoring is misleading in a child on emicizumab, and anti-emicizumab antibodies can rarely develop and reduce efficacy. [9] [12]
The avoidable pitfalls are well defined. The first is mistaking mild haemophilia A for von Willebrand disease because the von Willebrand factor was not measured — always send the antigen and ristocetin cofactor activity alongside the factor VIII. The second is delaying factor for a suspected intracranial bleed while waiting for imaging or a level. The third is failing to screen for inhibitors after each new exposure, so a child presents with uncontrolled bleeding from an undiagnosed inhibitor. The fourth is giving an intramuscular injection or a non-steroidal anti-inflammatory drug to a child with haemophilia. [1] [3]
Prognosis & Disposition
With modern comprehensive care, the life expectancy of a child with severe haemophilia now approaches that of the general population in high-income countries, and joint outcomes are transformed by early primary prophylaxis and emicizumab. A child diagnosed today and started on prophylaxis before the second joint bleed can expect near-normal joints, near-normal schooling and a near-normal lifespan, in stark contrast to the crippling arthropathy and early death of the pre-prophylaxis era. [4] [5]
The events that worsen prognosis are intracranial haemorrhage, the development of a high-titre inhibitor, and inadequate prophylaxis. Intracranial haemorrhage remains a leading cause of death and neurological disability despite modern care, and it is the reason every head injury is treated as a bleed first. A persistent high-responding inhibitor locks the child into bypassing agents and immune tolerance induction, with a higher bleeding rate and more complex management. [3] [7]
The disparity between high-income and resource-limited settings is stark. In countries without access to factor concentrate or emicizumab, severe haemophilia still causes crippling arthropathy, school dropout and death in childhood or early adulthood, and the World Federation of Hemophilia's humanitarian aid programme exists to narrow this gap. This global inequity is an examinable point and a reminder that the excellent prognosis of haemophilia is a product of access to treatment, not of the disease itself. [3]
Disposition is through a lifelong comprehensive haemophilia treatment centre that coordinates haematology, nursing, physiotherapy, social work and genetic counselling. The child and family learn home therapy and self-infusion, carry a medic alert device and an individualised bleed management plan, and have a clear safety-net for any bleed. Transition to adult care is structured and deliberate, handing over the prophylaxis regimen, inhibitor status, venous access, psychosocial needs and reproductive counselling. [1] [12]
Special Populations
The neonate of a known carrier mother is the population where prevention matters most. Plan the delivery to avoid instrumental intervention — ventouse and forceps carry a risk of intracranial and extracranial haemorrhage, and a caesarean section is not routinely required but instrumental delivery is avoided. Cord blood factor assays establish the diagnosis at birth. Defer circumcision until haemophilia is excluded, because circumcision bleeding is a common neonatal presentation. Any neonate with a cephalohaematoma, unexplained irritability or a poor feed is imaged for intracranial bleed. [3] [1]
The carrier female deserves attention because she is not always asymptomatic. Symptomatic carriers, with a factor level under 0.40 IU per mL from skewed X-inactivation (lyonisation), can bleed with surgery, dental work or menorrhagia, and a small number have bleeding severe enough to need prophylaxis. Genetic counselling covers the 50 percent chance of an affected son and a carrier daughter, prenatal and preimplantation genetic diagnosis, and reproductive choice. Measure the factor level in known and potential carriers, because the level, not the carrier status alone, determines bleeding risk. [2] [3]
The pregnant carrier mother needs a multidisciplinary plan involving obstetrics, haematology and the paediatric team. Delivery avoids instrumental intervention, cord blood is sent for factor assays, and the neonate is observed for bleeding. The transitioning adolescent needs structured handover of prophylaxis, inhibitor status, venous access, psychosocial support and reproductive counselling, because the transition to adult care is a high-risk period for disengagement and bleeding. [12] [4]
[2] [3]Evidence, Guidelines & Regional Differences
The cornerstone guidance is the World Federation of Hemophilia 2020 Guidelines for the Management of Hemophilia (3rd edition), which sets the factor-level severity thresholds, the prophylaxis regimens, the inhibitor definitions and the perioperative management used worldwide, and is reinforced by the 2024 ISTH clinical practice guideline for congenital haemophilia A and B built on the GRADE methodology. The SIPPET trial (Peyvandi 2016) and the RODIN cohort (Gouw 2013) shaped the debate on recombinant versus plasma-derived factor VIII and inhibitor risk, although practice varies and most high-income settings now use extended half-life and recombinant products. [1] [12] [8] [7]
The emicizumab HAVEN programme is the evidence that transformed haemophilia A prophylaxis. HAVEN 1 (Oldenburg 2017) showed that emicizumab prophylaxis in adults and adolescents with inhibitors reduced the annualised bleed rate by about 87 percent, and HAVEN 3 (Mahlangu 2018) showed the same benefit in patients without inhibitors. The paediatric HAVEN 2 study (Young 2019) confirmed efficacy and an acceptable subcutaneous route in children under twelve, including infants, and the HAVEN 7 study extended prophylaxis to the youngest infants. These trials established emicizumab as first-line prophylaxis for haemophilia A in many centres. [9] [10] [11]
The frontier of haemophilia therapy is moving fast. Extended half-life factor VIII and factor IX concentrates have reduced infusion frequency. Efanesoctocog alfa, an ultra-long-acting factor VIII, sustains near-normal trough levels with weekly dosing. Fitusiran, an antithrombin-targeting small interfering RNA, works for both haemophilia A and B with and without inhibitors. Gene therapy — valoctocogene roxaparvovec for haemophilia A and etranacogene dezaparvovec for haemophilia B — offers the prospect of a functional cure in adults, though paediatric use remains investigational. These advances are reshaping the long-term outlook but do not yet replace prophylaxis in children. [4] [12]
Exam Pearls
Haemophilia A is factor VIII (F8, Xq28) and haemophilia B is factor IX (F9, Xq27); both are X-linked recessive and share an identical severity scale defined by the factor level. The activated partial thromboplastin time is prolonged with a normal platelet count, prothrombin time and bleeding time, the mixing study corrects, and von Willebrand disease must always be excluded before diagnosing mild haemophilia A. [2] [3]
Severity is the central number: severe under 1 percent, moderate 1 to 5 percent, mild over 5 to 40 percent. The factor recovery rule is the central dose calculation: one IU per kg of factor VIII raises the level by about 2 percent, and one IU per kg of factor IX raises it by about 1 percent. Use it to dose for the target — 20 to 40 percent for a joint, 30 to 50 percent for a muscle, near 100 percent for a head bleed or surgery. [1] [4]
Inhibitors develop in about 30 percent of severe haemophilia A and 3 percent of severe haemophilia B, and a significant titre is at or above 0.6 Bethesda units. Emicizumab is the bispecific antibody that mimics factor VIIIa, given subcutaneously, transforming prophylaxis for haemophilia A with and without inhibitors. It does not treat haemophilia B. Any head injury is an intracranial bleed until proven otherwise — give factor first, then image. [7] [9]
The high-yield one-liners an examiner rewards: the commonest cause of severe haemophilia A is the intron 22 inversion; the bleeding pattern is delayed and deep (joint and muscle) because primary haemostasis is intact; never give an intramuscular injection or a non-steroidal anti-inflammatory to a child with haemophilia; the single most important mimic of mild haemophilia A is von Willebrand disease, excluded by the VWF antigen and ristocetin cofactor activity; and the paediatric HAVEN 2 study established subcutaneous emicizumab for infants and young children. [2] [3] [11]
References
- [1]Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia, 2020.PMID 32744769
- [2]Mannucci PM, Tuddenham EG The hemophilias--from royal genes to gene therapy. N Engl J Med, 2001.PMID 11396445
- [3]Peyvandi F, Garagiola I, Young G The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet, 2016.PMID 26897598
- [4]Berntorp E, Shapiro AD Modern haemophilia care. Lancet, 2012.PMID 22456059
- [5]Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med, 2007.PMID 17687129
- [6]Gringeri A, Lundin B, von Mackensen S, et al. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost, 2011.PMID 21255253
- [7]Gouw SC, van der Bom JG, Marijke van den Berg H Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med, 2013.PMID 23323899
- [8]Peyvandi F, Mannucci PM, Garagiola I, et al. A Randomized Trial of Factor VIII and Neutralizing Antibodies in Hemophilia A. N Engl J Med, 2016.PMID 27223147
- [9]Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N Engl J Med, 2017.PMID 28691557
- [10]Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab Prophylaxis in Patients Who Have Hemophilia A without Inhibitors. N Engl J Med, 2018.PMID 30157389
- [11]Young G, Sidonio RF, Liesner R, et al. A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood, 2019.PMID 31697801
- [12]Rezende SM, Cogo PE, Hsu TC, et al. International Society on Thrombosis and Haemostasis clinical practice guideline for treatment of congenital hemophilia A and B based on the Grading of Recommendations Assessment, Development, and Evaluation methodology. J Thromb Haemost, 2024.PMID 39043543