Paeds · haematology-oncology-and-transfusion
Hereditary spherocytosis and membrane disorders
Also known as Hereditary spherocytosis · HS · Hereditary elliptocytosis · Hereditary pyropoikilocytosis · Hereditary stomatocytosis · Red cell membrane disorders
Fellowship guide to hereditary spherocytosis and the inherited red cell membrane disorders: the pale jaundiced child with splenomegaly, the spherocyte on the blood film, the eosin-5-maleimide binding test, the severity spectrum from compensated to transfusion-dependent, folate supplementation and transfusion support, splenectomy timing and technique (total versus partial), the pre-splenectomy vaccination and post-splenectomy antibiotic prophylaxis bundle, the post-splenectomy sepsis risk, and the membrane disorders that must not be splenectomised (hereditary stomatocytosis).
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the six-year-old who has always been a little pale and yellow, sent to you because the teacher noticed she tires easily, and whose blood film shows small dense round cells without central pallor scattered among otherwise normal red cells. Her mother had her gallbladder out at twenty-five and her grandmother was always told she had a fragile blood condition. That family story is hereditary spherocytosis, and the blood film has just handed you the diagnosis. [1]
Hereditary spherocytosis is a group of inherited disorders of the red blood cell membrane in which a quantitative or qualitative deficiency of one or more membrane skeleton proteins destabilises the vertical interactions that tether the lipid bilayer to the underlying spectrin-based cytoskeleton. The membrane blebs off in small fragments, the cell loses surface area without losing volume, and the resulting decrease in the surface-to-volume ratio forces the biconcave disc into a sphere. The spherocyte is rigid and lacks the deformability to pass through the splenic cords, so it is trapped and destroyed by splenic macrophages — the extravascular haemolysis that defines the clinical picture. [1] [4]
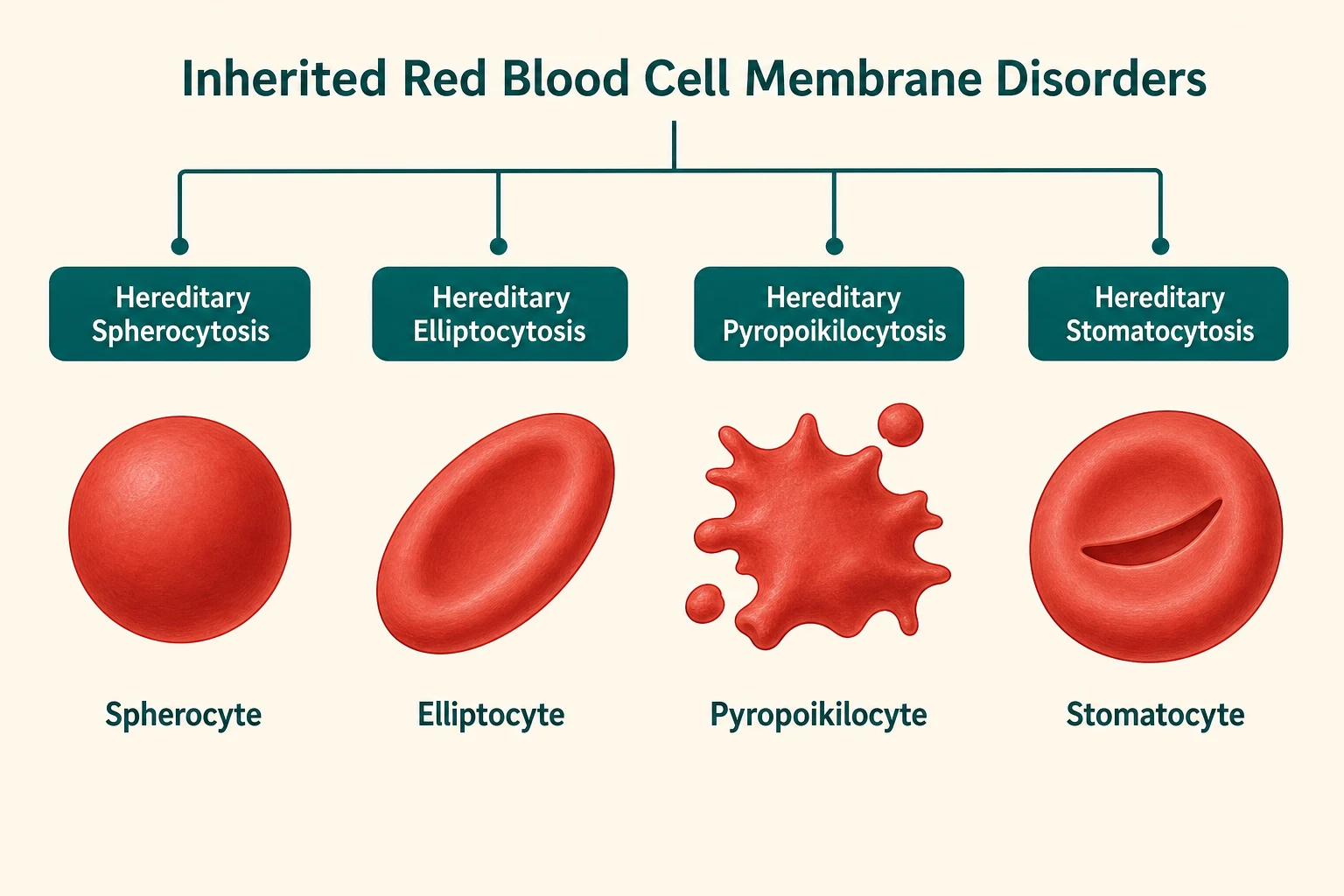
The inherited red cell membrane disorders form a family defined by which protein interaction is defective and what shape the cell takes. Hereditary spherocytosis, the commonest and most examinable, is a vertical interaction defect producing spherocytes. Hereditary elliptocytosis is a horizontal skeleton defect producing elliptocytes, usually mild or asymptomatic. Hereditary pyropoikilocytosis is the severe neonatal form. Hereditary stomatocytosis is a cation-leak disorder producing stomatocytes, and it carries the critical caveat that splenectomy is dangerous. Together these disorders share the themes of inherited haemolysis, membrane morphology on the blood film, and splenic destruction — but they diverge sharply in management, which is why distinguishing them matters enormously. [3] [4]
Classification
The most useful classification of hereditary spherocytosis runs along three axes that each change the clinical approach: the clinical severity, the molecular defect, and the broader family of membrane disorders it belongs to. [2]
On the severity axis, the British Society for Haematology classifies hereditary spherocytosis into mild, moderate and severe disease based on the haemoglobin, the reticulocyte count and the bilirubin. Mild disease is asymptomatic or minimally symptomatic with a haemoglobin above 100 g per litre, modestly raised reticulocytes and bilirubin, and compensated haemolysis that may never need intervention. Moderate disease has a haemoglobin of 80 to 110 g per litre with intermittent symptoms and occasional transfusion need. Severe disease has a haemoglobin below 80 g per litre, marked reticulocytosis and hyperbilirubinaemia, and is transfusion-dependent until splenectomy. These thresholds guide the decision to offer splenectomy, because mild disease can be managed conservatively while moderate and severe disease usually warrant surgery. [2] [1]
The severity spectrum of hereditary spherocytosis
On the molecular axis, the defect lies in one of the proteins of the vertical linkage complex. The most common is ankyrin deficiency, accounting for roughly half of cases, followed by beta-spectrin deficiency, band 3 deficiency, alpha-spectrin deficiency, and protein 4.2 deficiency. The inheritance is autosomal dominant in about seventy-five per cent of cases, with the remainder autosomal recessive or arising de novo. The severity correlates loosely with the degree of membrane protein deficiency, and molecular testing is available but not required for most clinical decisions. [1] [6]
On the broader family axis, hereditary elliptocytosis is caused by defects in the horizontal skeletal interactions, most commonly alpha-spectrin mutations impairing spectrin dimer self-association, and it produces elliptocytes that are usually stable enough to cause little or no haemolysis. Hereditary pyropoikilocytosis is the severe neonatal form, typically a compound heterozygote for an elliptocytosis mutation and a low-expression alpha-spectrin allele, presenting with severe haemolysis and bizarre fragmented cells. Southeast Asian ovalocytosis is a rigid band 3 variant that is clinically mild and famously protective against cerebral malaria. [3] [4]
Epidemiology & Risk Factors
Hereditary spherocytosis is the commonest inherited haemolytic anaemia in people of Northern European descent, with an estimated prevalence of about one in two thousand. The true prevalence may be higher, because mild cases are underdiagnosed throughout life. It is less common but well recognised in other populations, and it is found worldwide wherever the gene pool carries the relevant mutations. [1] [2]
The inheritance pattern shapes the family history. About three-quarters of cases are autosomal dominant, which means an affected parent is the rule rather than the exception and the family history is often the first clue. The remaining quarter are autosomal recessive or arise from de novo mutations, and these tend to present with more severe disease because the child inherits two defective alleles or has a dominant-negative mutation from one parent. A negative family history does not exclude hereditary spherocytosis, and a high index of suspicion is warranted in any child with unexplained haemolysis. [1] [4]
The most important risk factor for a poor outcome is not the genotype itself but a delayed or missed diagnosis. Neonates with severe hereditary spherocytosis can develop bilirubin encephalopathy if the jaundice is attributed to physiological causes or ABO incompatibility without investigation. Children who are undiagnosed are at risk of aplastic crisis, gallstone complications and severe anaemic episodes, and they miss out on folic acid supplementation, transfusion support and timely splenectomy planning. [5]
Pathophysiology
The central mechanism of hereditary spherocytosis is a defect in the vertical interactions that link the red cell membrane lipid bilayer to the underlying spectrin-based cytoskeleton. When these linkages are weakened by a deficiency of ankyrin, spectrin, band 3 or protein 4.2, the lipid bilayer is no longer firmly tethered to the skeleton. The unsupported membrane blebs off in small vesicles, the cell loses surface area without losing haemoglobin content or volume, and the surface-to-volume ratio falls. A normal biconcave disc has a large excess of surface area over volume, which is what gives it its shape and its remarkable deformability. Remove that excess and the cell is forced into a sphere — the least surface area for a given volume. [1] [4]
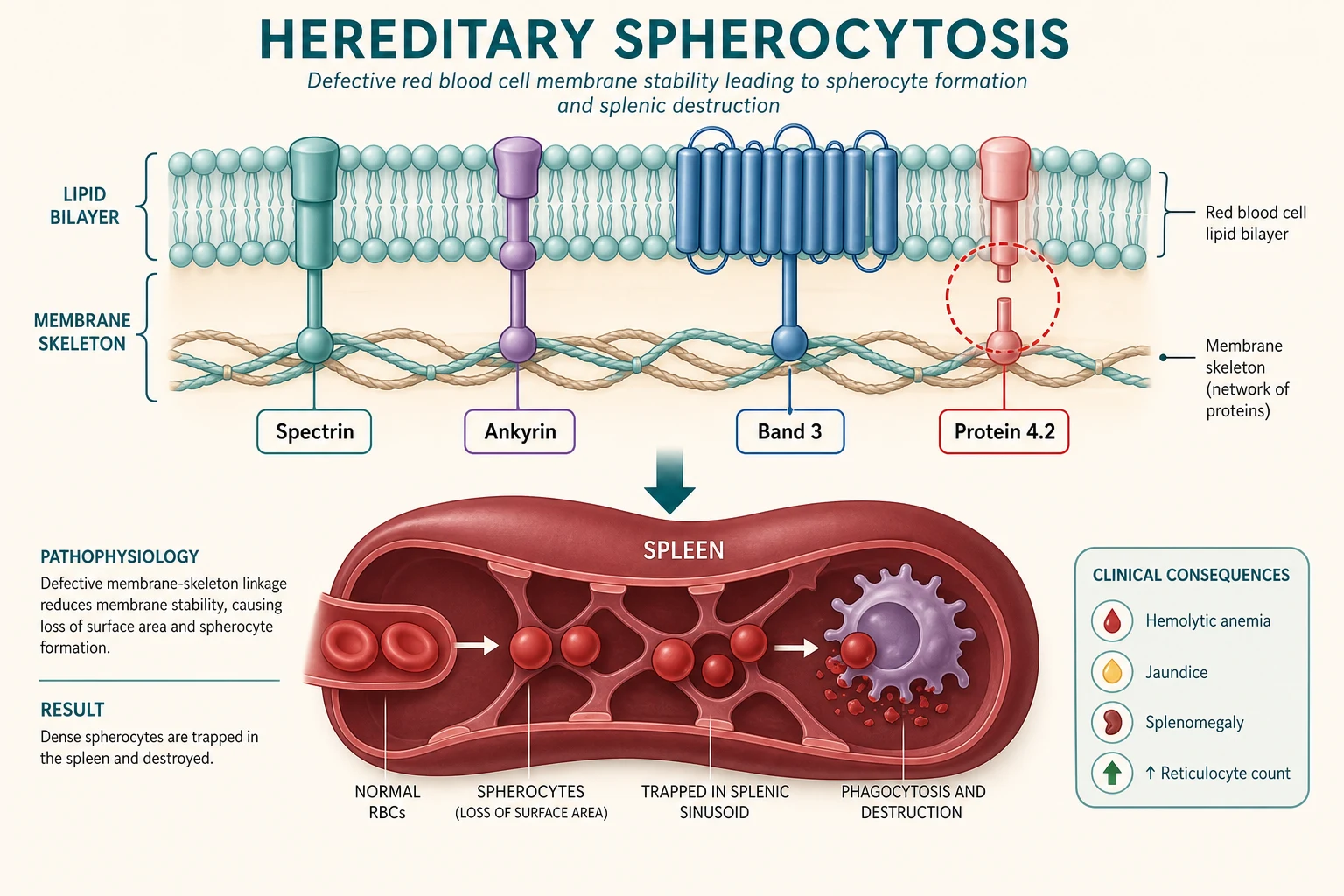
The spherocyte that results is mechanically unstable and rigid. A normal red cell deforms to pass through the narrow slits of the splenic cords, but the spherocyte, with its reduced surface area and reduced deformability, cannot. It is trapped in the splenic red pulp, where splenic macrophages and the harsh metabolic environment of low glucose and low pH accelerate its destruction. This extravascular haemolysis is the engine of the anaemia, jaundice and splenomegaly that define the clinical picture, and it is also why splenectomy is curative — removing the organ of destruction stops the haemolysis even though it does not correct the underlying membrane defect. [1]
The metabolic environment of the spleen is itself part of the pathology. Within the splenic cords, red cells are exposed to low glucose, acidic pH and abundant macrophages, and the already-compromised spherocyte is further damaged by repeated passages. This produces a self-perpetuating cycle in which the spleen both traps and worsens the spherocytes, and it explains why even moderately affected children improve dramatically after splenectomy. [4]
[1] [8]The broader membrane disorders share the theme of impaired mechanical stability but differ in the plane of the defect. Hereditary elliptocytosis is a horizontal skeleton defect: mutations in alpha-spectrin or beta-spectrin impair the self-association of spectrin dimers into tetramers, weakening the two-dimensional lattice that lies parallel to the membrane. The cell elongates into an ellipse under the shear stress of the circulation but is usually stable enough to survive, which is why most patients are asymptomatic or have only mild haemolysis. Hereditary pyropoikilocytosis inherits two defective alpha-spectrin alleles, producing such severe skeletal weakness that the cell fragments into bizarre shapes under circulatory stress. [3] [4]
Hereditary stomatocytosis is mechanistically distinct from all of the above. It is a cation-leak disorder — mutations in transporters such as PIEZO1 in dehydrated hereditary stomatocytosis produce an uncontrolled leak of potassium and sodium that alters intracellular hydration and produces a cell with a mouth-shaped central pallor. The leak is the disease, and the resulting cells are abnormally permeable. This distinction matters enormously for management, because splenectomy, which cures the other membrane disorders, is dangerous in stomatocytosis due to a high risk of fatal thromboembolism. [7] [6]
Clinical Presentation
The clinical presentation spans the full range from an incidental finding on a routine blood count to a life-threatening neonatal anaemia, and the severity is determined by the degree of membrane protein deficiency and the resulting rate of haemolysis. The three cardinal features are chronic or episodic haemolytic anaemia, jaundice and splenomegaly, but the child who walks in with all three prominently is the exception rather than the rule. [1] [2]
The anaemia is variable in severity. Mildly affected children may have a haemoglobin at or near the normal range, with only a slightly raised reticulocyte count and bilirubin, and they may never come to medical attention unless an intercurrent illness or a routine blood count reveals the spherocytes. Moderately affected children have a chronically low haemoglobin, easy fatigability, pallor and exercise intolerance, with intermittent exacerbations during viral infections. Severely affected children are transfusion-dependent and present in early infancy with profound anaemia. [4]
Jaundice reflects the accelerated breakdown of haemoglobin and the resulting bilirubin load on the liver. It is typically mild and fluctuating, worse during intercurrent infections, and it may be the first sign in a neonate who presents with prolonged or severe hyperbilirubinaemia. The chronic bilirubin load also predisposes to pigment gallstones, which can present as biliary colic or cholecystitis in adolescence or early adulthood — sometimes the first recognised manifestation of the disease in a family. [5] [1]
Splenomegaly is the hallmark physical sign and is present in the great majority of symptomatic children. The spleen is enlarged because it is the organ of red cell destruction, and it may reach several centimetres below the costal margin. The liver may be mildly enlarged as well, and frontal bossing and other skeletal changes of chronic marrow expansion are seen in severely affected children. Growth may be impaired in transfusion-dependent disease, and folic acid deficiency can develop because of the high turnover of haematopoietic cells. [4]
| Clinical picture | What it implies | Act |
|---|
The acute presentations that bring a child with hereditary spherocytosis to emergency attention are the aplastic crisis, the severe haemolytic exacerbation and, in the neonate, severe hyperbilirubinaemia. A parvovirus B19 aplastic crisis is the most dangerous: the virus infects erythroid precursors, transiently shutting down red cell production at a time when the child depends on a high reticulocyte count to compensate for ongoing haemolysis. The haemoglobin falls rapidly over days, the reticulocyte count collapses, and the child can become critically anaemic. [1] [5]
Differential Diagnosis
Build the differential diagnosis around the finding of a haemolytic anaemia with spherocytes on the blood film, because spherocytes are not unique to hereditary spherocytosis. The first and most urgent alternative to exclude is autoimmune haemolytic anaemia, which produces spherocytes that are morphologically indistinguishable from those of hereditary spherocytosis. The direct antiglobulin test is the decisive discriminator: it is positive in autoimmune haemolysis and negative in hereditary spherocytosis. [2] [4]
The second layer is the other inherited haemolytic anaemias. G6PD deficiency produces episodic haemolysis with bite cells and blister cells rather than spherocytes, and it is X-linked. Pyruvate kinase deficiency produces a chronic non-spherocytic haemolytic anaemia with echinocytes. The haemoglobinopathies — thalassaemia and sickle cell disease — have their own characteristic film appearances and are distinguished by haemoglobin electrophoresis. Congenital dyserythropoietic anaemia is a rarer mimic that can produce a false-positive EMA binding test and should be considered when the film and clinical picture are atypical. [3] [2]
Autoimmune haemolysis
DAT positive
- Spherocytes identical to HS
- Direct antiglobulin test positive
- Acute onset, no family history
- Treat with steroids, not splenectomy first
Other enzymopathies
- G6PD: bite cells, episodic
- Pyruvate kinase: echinocytes, chronic
- Enzyme assay confirms
- X-linked or autosomal recessive
Haemoglobinopathies
- Thalassaemia: target cells
- Sickle cell: sickle cells
- Haemoglobin electrophoresis
- More common in malaria-endemic regions
Stomatocytosis
do not splenectomise
- Stomatocytes on film
- Cation leak on osmotic gradient
- Splenectomy contraindicated
- Thromboembolism risk
The third layer is the neonatal differential. Neonatal jaundice with anaemia has a broad differential that includes ABO and Rh haemolytic disease of the newborn, G6PD deficiency, congenital infections and hereditary spherocytosis. The family history, the direct antiglobulin test, the blood film and the maternal and infant blood groups help separate them. Hereditary spherocytosis should be considered in any neonate with early-onset, severe or prolonged jaundice that is disproportionate to the apparent cause, because the consequences of missing severe neonatal disease are bilirubin encephalopathy and kernicterus. [5]
The decisive step when the diagnosis is uncertain is a synthesis of the blood film, the EMA binding test, the family history and, where needed, further specialised testing such as osmotic gradient ektacytometry or molecular analysis. The direct antiglobulin test should always be performed to exclude autoimmune haemolysis before attributing spherocytes to an inherited cause. [2] [6]
Clinical & Bedside Assessment
Assessment begins with the history, where the family story is often the single most useful piece of information. Ask about a family history of anaemia, jaundice, gallstones, splenectomy or cholecystectomy, because the autosomal dominant inheritance means an affected parent, grandparent or sibling is common. Ask about the onset and pattern of pallor, jaundice and exercise intolerance, and about any episodes of dark urine, abdominal pain or severe illness that might represent a haemolytic or aplastic crisis. Document the neonatal history, because severe neonatal jaundice requiring phototherapy or exchange transfusion is a frequent early clue that was attributed to another cause. [1] [5]
The abdominal examination is the core of the bedside assessment. Palpate carefully for splenomegaly, which is present in most symptomatic children and may be the first finding on a routine examination. Assess the liver size, look for pallor and scleral icterus, and examine for frontal bossing and other signs of chronic marrow expansion in severely affected children. Document the growth parameters, because chronic anaemia can impair growth, and assess pubertal development. [4]
[1] [5]A careful assessment of the severity at presentation shapes the entire management plan. Document the baseline haemoglobin, reticulocyte count and bilirubin, because the trend of these values over time determines whether the child can be managed conservatively with folate alone or needs transfusion support and consideration of splenectomy. Ask about gallbladder symptoms, because the chronic bilirubin load produces pigment gallstones that may complicate the course. Assess for any features that suggest a different diagnosis, such as a positive family history of stomatocytosis or a blood film with unusual cell shapes. [2]
Investigations
The investigation strategy serves three purposes: to confirm the diagnosis of hereditary spherocytosis, to assess the severity and complications, and to exclude the mimics. The full blood count shows a variable anaemia with a raised mean cell haemoglobin concentration, because the spherocyte is dense and has lost surface area, and the reticulocyte count is elevated, reflecting the marrow response to chronic haemolysis. The blood film is the cornerstone of the morphological diagnosis, showing spherocytes — small, dense, deeply staining cells with no central pallor — in variable numbers alongside polychromatic reticulocytes. [2] [4]
The bilirubin is raised, predominantly the unconjugated fraction, and the lactate dehydrogenase is elevated, reflecting red cell destruction. Haptoglobin is low or absent. These are the standard markers of haemolysis, and their degree correlates with the severity of the disease. The direct antiglobulin test must be performed and must be negative before the diagnosis of hereditary spherocytosis is accepted, because autoimmune haemolysis produces identical spherocytes and is excluded only by this test. [2]
The eosin-5-maleimide binding test has become the first-line confirmatory investigation in most centres. Eosin-5-maleimide is a fluorescent dye that binds to band 3 and associated proteins on the red cell membrane. In hereditary spherocytosis, the reduced band 3 content means less dye binds, and flow cytometry shows reduced fluorescence compared with control cells. The test has a sensitivity of about ninety-three per cent and a specificity of about ninety-eight per cent, it is rapid, requires only a small blood sample, and it can be performed on the same day as the blood film. It does not distinguish hereditary spherocytosis from hereditary pyropoikilocytosis or some cases of congenital dyserythropoietic anaemia, so the result must be interpreted alongside the blood film and the clinical context. [2] [6]
The standard diagnostic workup
Full blood count with mean cell haemoglobin concentration, reticulocyte count and blood film.
Unconjugated bilirubin, lactate dehydrogenase and haptoglobin to confirm haemolysis.
Direct antiglobulin test to exclude autoimmune haemolytic anaemia.
Eosin-5-maleimide binding test by flow cytometry as the first-line confirmatory test.
Abdominal ultrasound for splenomegaly and gallstones.
Family screening with blood count, film and EMA test for first-degree relatives.
Consider osmotic gradient ektacytometry or molecular testing for atypical cases.
The osmotic fragility test, historically the standard confirmatory test, has been largely superseded by the EMA binding test because it is less sensitive, particularly in mild cases, and requires fresh blood. It remains useful as a second-line test and in settings where flow cytometry is unavailable. Osmotic gradient ektacytometry is a specialised technique available in reference laboratories that can distinguish hereditary spherocytosis from hereditary stomatocytosis and other membrane disorders by measuring the deformability of red cells across an osmotic gradient, and it is particularly useful in atypical or complex cases. [2] [3]
Abdominal ultrasound should be performed to document the spleen size and to screen for gallstones, because pigment cholelithiasis is a common complication of chronic haemolysis and may influence the timing of surgery. If splenectomy is being planned and gallstones are present, concomitant cholecystectomy is often performed in the same operation. [2]
Management — Resuscitation
Resuscitation in hereditary spherocytosis is rarely about airway and circulation in the routine case, because most children are chronically stable or mildly symptomatic. The two situations that demand urgent resuscitation are the parvovirus B19 aplastic crisis and severe neonatal anaemia. In an aplastic crisis, the haemoglobin can fall rapidly to life-threatening levels, and the child presents with pallor, tachycardia, tachypnoea and signs of cardiovascular compromise. The treatment is urgent transfusion of packed red cells, because the marrow is temporarily unable to produce red cells and the haemolysis is ongoing. [1] [5]
SPHERES
In the neonate with severe hereditary spherocytosis, the emergency is severe hyperbilirubinaemia that threatens the developing brain. Phototherapy is the first step, but exchange transfusion may be required if the bilirubin rises to dangerous levels or if the anaemia is severe. The principle is to prevent bilirubin encephalopathy and kernicterus while confirming the diagnosis and planning ongoing care. [5]
For the child with a severe haemolytic exacerbation triggered by an intercurrent infection, supportive care with transfusion, hydration and treatment of the underlying infection is usually sufficient. These episodes are self-limited, and the child returns to their baseline once the infection resolves. [1]
Management — Definitive & Stepwise
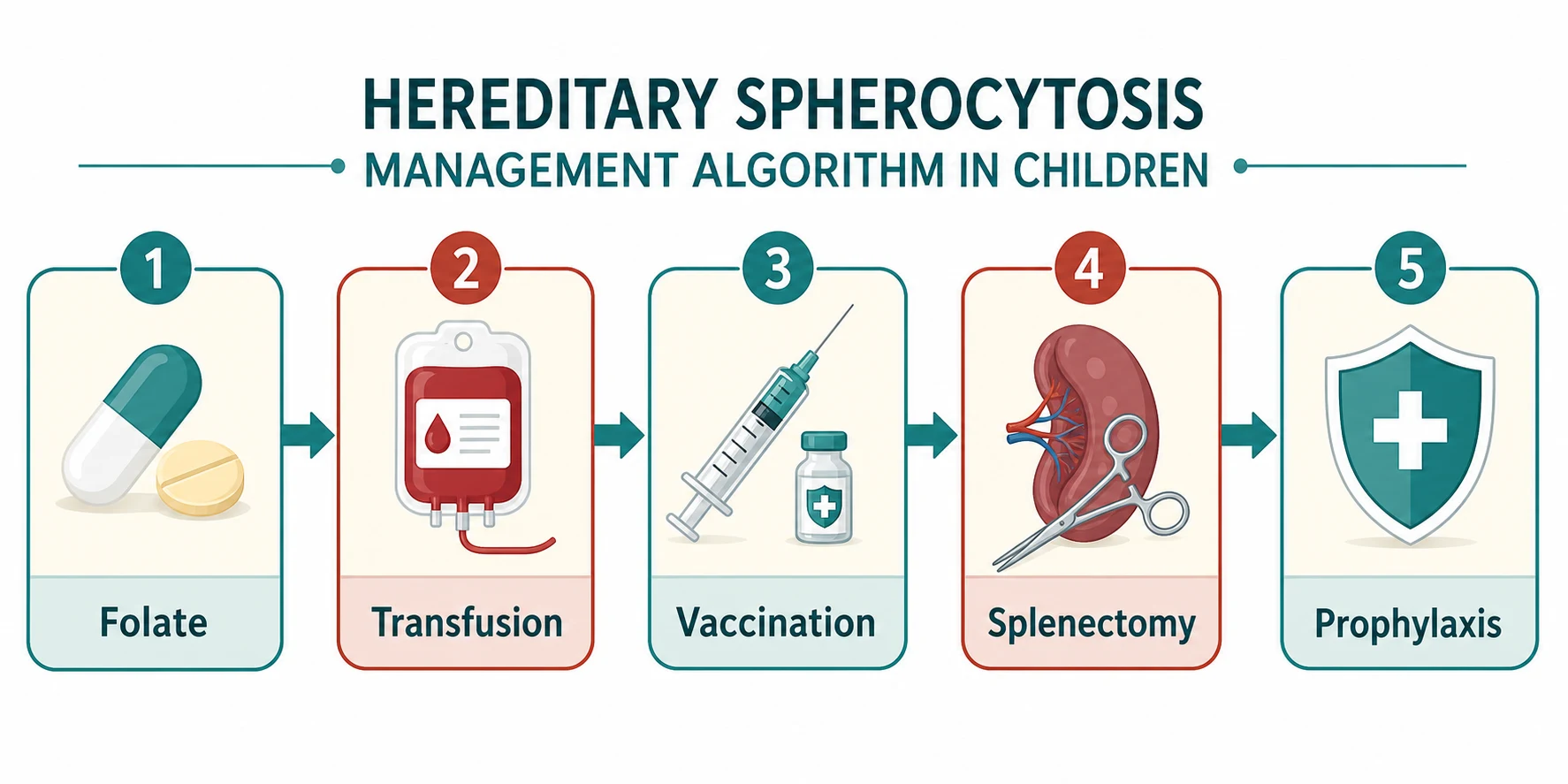
The definitive management of hereditary spherocytosis is layered, moving from conservative support through transfusion to splenectomy, and the step taken depends on the severity and the age of the child. Every child with hereditary spherocytosis, regardless of severity, should receive folic acid supplementation to support the expanded erythropoiesis and prevent megaloblastic crisis. The standard dose is folic acid 5 mg daily in older children and adults, and 2.5 mg daily in younger children. [2]
The treatment arc of hereditary spherocytosis
For children with moderate or severe disease, transfusion support is part of routine management until splenectomy can be performed. Transfusions are given for symptomatic anaemia, during aplastic crises, and perioperatively. The transfusion threshold depends on the clinical context: a haemoglobin below 70 g per litre with symptoms, or below 60 g per litre in a neonate with severe disease, typically warrants transfusion. In the chronically transfused child, iron chelation may be needed to prevent iron overload, though this is uncommon because splenectomy usually eliminates the transfusion requirement. [1] [2]
Splenectomy is the only curative treatment for the haemolysis of hereditary spherocytosis. It removes the organ of red cell destruction, so the haemolysis resolves, the haemoglobin normalises and the jaundice clears. The indications are moderate to severe disease with symptomatic haemolysis, transfusion dependence, or recurrent haemolytic or aplastic crises. The British Society for Haematology guideline recommends splenectomy for severe disease and selectively for moderate disease, and it emphasises that the decision must balance the benefit of correcting the haemolysis against the lifelong risk of overwhelming post-splenectomy infection. [2] [8]
Phenoxymethylpenicillin (penicillin V) — post-splenectomy prophylaxis
The timing of splenectomy is a critical decision. The overwhelming post-splenectomy infection risk is highest in young children, so splenectomy should be deferred until at least six years of age wherever possible. In a child with severe transfusion-dependent disease who cannot wait, a partial splenectomy may be considered as a temporising measure to reduce the haemolysis while preserving some splenic immune function. The laparoscopic approach is preferred for both total and partial splenectomy, with shorter recovery and fewer complications than open surgery. [8] [9]
The choice between total and partial splenectomy has been the subject of debate and recent systematic review. Total splenectomy eliminates the haemolysis completely and prevents recurrence, but it removes all splenic immune function and carries the full risk of overwhelming post-splenectomy infection. Partial splenectomy preserves some immune function and may reduce the infection risk, but a recent meta-analysis found that it carries a higher rate of reoperation and recurrence of haemolysis. The current consensus favours total splenectomy for definitive cure, with partial splenectomy reserved for children under six years with severe disease who cannot wait, or for families who prioritise immune preservation after full counselling. [9] [8]
Specific Subtypes & Scenarios
Neonatal hereditary spherocytosis is the presentation that demands the most vigilance. The neonate may present with early-onset, severe or prolonged jaundice, a falling haemoglobin, or both, and the diagnosis is easily missed because the differential of neonatal jaundice is broad and hereditary spherocytosis is not always considered. Severe neonatal disease can cause bilirubin encephalopathy if the jaundice is undertreated. The approach is to investigate any neonate with disproportionate or prolonged jaundice with a blood count, film, reticulocyte count, bilirubin fractionation and direct antiglobulin test, and to perform an EMA binding test if haemolysis is confirmed. Phototherapy and exchange transfusion are used as needed, and the family is screened. [5]
The parvovirus B19 aplastic crisis is the acute scenario that every clinician caring for a child with hereditary spherocytosis must be prepared for. Parvovirus B19 infects erythroid progenitor cells via the P antigen and transiently halts red cell production. In a child whose survival depends on a high reticulocyte count to compensate for ongoing haemolysis, this precipitous drop in production causes a rapid and severe fall in haemoglobin. The reticulocyte count collapses, and the child presents with pallor, fatigue and cardiovascular compromise over days. The treatment is urgent transfusion, and the child should be isolated from pregnant contacts because parvovirus B19 can cause hydrops fetalis. Recovery is heralded by a brisk reticulocytosis as the marrow recovers. [1] [5]
Hereditary elliptocytosis and hereditary pyropoikilocytosis are the horizontal skeleton disorders. Hereditary elliptocytosis is usually mild and asymptomatic, requiring no treatment beyond folic acid if there is any haemolysis. The common form is autosomal dominant, prevalent in malaria-endemic regions, and most patients are detected incidentally on a blood film showing more than twenty-five per cent elliptocytes. Hereditary pyropoikilocytosis is the severe neonatal form, presenting with severe haemolysis and bizarre fragmented cells, and it requires transfusion support and often splenectomy in early childhood. [3] [4]
Hereditary stomatocytosis is the membrane disorder that must not be splenectomised. It is a cation-leak disorder, either overhydrated or dehydrated (also called xerocytosis), and it produces a chronic haemolytic anaemia with stomatocytes on the blood film. The critical point is that splenectomy in hereditary stomatocytosis carries a high risk of thromboembolism, including fatal pulmonary hypertension and venous thrombosis, because the abnormally permeable cells are not fully cleared and contribute to a prothrombotic state. Before any splenectomy for a presumed membrane disorder, the diagnosis must be confirmed and stomatocytosis excluded, ideally by osmotic gradient ektacytometry or molecular testing. [7] [6]
Complications & Pitfalls
The complications of hereditary spherocytosis divide into those of the disease itself and those of its treatment. The disease complications include gallstones from the chronic bilirubin load, growth impairment from chronic anaemia, folic acid deficiency from the expanded erythropoiesis, and the acute crises — aplastic, haemolytic and severe neonatal jaundice. Pigment gallstones are found in a substantial proportion of adults with hereditary spherocytosis and can present as biliary colic or cholecystitis, sometimes as the first manifestation of the disease. [1] [4]
The risks that drive management
The treatment complications are dominated by the consequences of splenectomy. Overwhelming post-splenectomy infection is the most feared: the asplenic child cannot clear encapsulated organisms such as Streptococcus pneumoniae, Neisseria meningitidis and Haemophilus influenzae type b, and a previously well child can develop fulminant sepsis and die within hours. The lifetime risk is low but the case fatality is high, which is why the pre-splenectomy vaccination bundle and post-splenectomy antibiotic prophylaxis are mandatory and lifelong. The risk is highest in young children and in the first two years after splenectomy, which is why the surgery is deferred until at least six years wherever possible. [10] [8]
Thrombocytosis is common after splenectomy and is usually transient, but it can contribute to a prothrombotic state, particularly in children who undergo splenectomy for the wrong diagnosis — most dangerously hereditary stomatocytosis. This is why excluding stomatocytosis before splenectomy is a non-negotiable safety step. Pulmonary hypertension is a recognised long-term complication of asplenia, thought to relate to altered clearance of particulate matter from the circulation. [7]
The avoidable pitfalls are clinical and systemic. Failing to investigate unexplained neonatal jaundice with anaemia misses severe hereditary spherocytosis. Failing to perform a direct antiglobulin test before diagnosing hereditary spherocytosis misdiagnoses autoimmune haemolysis, which needs immunosuppression rather than splenectomy. Splenectomising a child with hereditary stomatocytosis, mistaking it for hereditary spherocytosis, can cause fatal thromboembolism. And performing splenectomy without completing the vaccination and prophylaxis bundle exposes the child to preventable overwhelming post-splenectomy infection. [2] [7]
[2]Prognosis & Disposition
The prognosis of hereditary spherocytosis is excellent when the diagnosis is made and the condition is managed appropriately. Children with mild disease lead entirely normal lives with folic acid supplementation and no other intervention, and their life expectancy is normal. Children with moderate or severe disease who undergo splenectomy are cured of their haemolysis and return to a normal haemoglobin, though they carry the lifelong responsibility of asplenic precautions. [1] [4]
The prognosis after splenectomy is shaped by the asplenic state. The haemoglobin normalises, the jaundice clears, and the transfusion requirement disappears, but the child is permanently at risk of overwhelming post-splenectomy infection. With appropriate vaccination, antibiotic prophylaxis, and a febrile-illness action plan, this risk is substantially mitigated, and most splenectomised children live full and active lives. The ongoing surveillance focuses on reinforcing the asplenic precautions, monitoring for late complications such as pulmonary hypertension, and ensuring that the vaccination schedule is kept up to date through adolescence and into adulthood. [10] [8]
Disposition after splenectomy includes education of the family and the child about the asplenic state, provision of a medic alert device and a patient-held information card, a clear febrile-illness action plan that instructs the family to seek immediate medical attention for any fever, and a plan for regular vaccination boosters. Transition to adult care in adolescence should include a formal handover of the asplenic precautions, because the risk does not diminish with age. [2] [10]
Special Populations
Neonates and infants deserve special attention because the presentation can be dramatic and the diagnosis is often missed. Severe neonatal hereditary spherocytosis can cause bilirubin encephalopathy, and any neonate with disproportionate or prolonged jaundice and a falling haemoglobin should be investigated for haemolysis with a blood film, reticulocyte count, bilirubin fractionation, direct antiglobulin test and EMA binding test. Family screening of both parents is essential, because the autosomal dominant inheritance means one parent is usually affected. [5]
Children of Indigenous, Pacific and migrant heritage may carry membrane variants that are less common in the Northern European population and may be under-recognised. South-East Asian ovalocytosis is prevalent in parts of Papua New Guinea, Indonesia and the Philippines and is often detected incidentally. Culturally safe assessment, attention to language and health literacy, and a low threshold to investigate unexplained haemolysis are part of standard care. Access to specialist haematology services and the cost of investigations and surgery can be barriers for socioeconomically disadvantaged families, and the role of telehealth and retrieval services is important in rural and remote settings. [3]
The adolescent transitioning to adult care with hereditary spherocytosis is a defined population that needs structured handover. Whether the adolescent has had a splenectomy or is managed conservatively, the transition should include a full review of the diagnosis and management plan, reinforcement of the asplenic precautions if applicable, a plan for reproductive health counselling (because hereditary spherocytosis is inherited), and a warm handover to an adult haematologist. The risk of lost follow-up, non-adherence with prophylaxis, and unplanned presentations is highest in this age group, and structured transition programmes address these risks. [1] [10]
The child with hereditary stomatocytosis is the special population that must never be splenectomised. The cation-leak disorders — overhydrated and dehydrated hereditary stomatocytosis — produce chronic haemolysis that resembles hereditary spherocytosis, but splenectomy carries a high risk of fatal thromboembolism. These children are managed conservatively with folic acid and transfusion support, and novel therapies targeting the cation leak are under investigation. The key clinical message is to confirm the diagnosis and exclude stomatocytosis before any splenectomy for a presumed membrane disorder. [7] [6]
Evidence, Guidelines & Regional Differences
| Region | Key guideline | Screening test | Splenectomy approach |
|---|
The two areas of evolving evidence and controversy that a fellowship candidate should be able to discuss are the choice between total and partial splenectomy, and the management of hereditary stomatocytosis. The systematic review comparing partial and total splenectomy found that total splenectomy provides superior haematological outcomes but at the cost of complete loss of splenic immune function, while partial splenectomy preserves some immune function but carries a higher rate of reoperation. For hereditary stomatocytosis, the evidence is clear that splenectomy is dangerous and should be avoided, but the optimal alternative management is still evolving, with novel therapies targeting the cation leak under investigation. [9] [7]
Exam Pearls
References
- [1]Perrotta S, Gallagher PG, Mohandas N Hereditary spherocytosis. Lancet, 2008.PMID 18940465
- [2]Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol, 2012.PMID 22055020
- [3]Da Costa L, Galimand J, Fenneteau O, et al. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev, 2013.PMID 23664421
- [4]Gallagher PG Abnormalities of the erythrocyte membrane. Pediatr Clin North Am, 2013.PMID 24237975
- [5]Christensen RD, Yaish HM, Gallagher PG A pediatrician's practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics, 2015.PMID 26009624
- [6]Iolascon A, Andolfo I, Russo R Advances in understanding the pathogenesis of red cell membrane disorders. Br J Haematol, 2019.PMID 31364155
- [7]Andolfo I, Russo R, Gambale A, et al. Hereditary stomatocytosis: An underdiagnosed condition. Am J Hematol, 2018.PMID 28971506
- [8]Casale M, Perrotta S Splenectomy for hereditary spherocytosis: complete, partial or not at all? Expert Rev Hematol, 2011.PMID 22077527
- [9]Tang X, Xue J, Zhang J, et al. The efficacy of partial versus total splenectomy in the treatment of hereditary spherocytosis in children: a systematic review and meta-analysis. Pediatr Surg Int, 2024.PMID 39470805
- [10]Liu Y, Jin S, Xu R, et al. Hereditary spherocytosis before and after splenectomy and risk of hospitalization for infection. Pediatr Res, 2023.PMID 35915237