AL Amyloidosis
AL amyloidosis (immunoglobulin light chain amyloidosis) is a rare, systemic protein misfolding disorder caused by the ex... MRCP exam preparation.
What matters first
AL amyloidosis (immunoglobulin light chain amyloidosis) is a rare, systemic protein misfolding disorder caused by the ex... MRCP exam preparation.
Cardiac involvement with elevated NT-proBNP less than 8500 ng/L (Stage IV disease)
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Cardiac involvement with elevated NT-proBNP less than 8500 ng/L (Stage IV disease)
- Rapidly progressive organ failure involving multiple systems
- Severe autonomic neuropathy with orthostatic hypotension
- Troponin T less than 0.06 ng/mL indicating cardiac amyloid
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- AA Amyloidosis
- ATTR Amyloidosis (Hereditary and Wild-Type)
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
AL Amyloidosis
1. Overview
AL amyloidosis (immunoglobulin light chain amyloidosis) is a rare, systemic protein misfolding disorder caused by the extracellular deposition of fibrillar aggregates derived from monoclonal immunoglobulin light chains. [1] The condition arises from an underlying clonal plasma cell or B-lymphocyte proliferative disorder that produces abnormal light chains (most commonly lambda), which misfold and aggregate into insoluble amyloid fibrils with characteristic β-pleated sheet configuration. [2] These fibrils deposit in various organs, leading to progressive organ dysfunction and ultimately organ failure.
AL amyloidosis is the most common form of systemic amyloidosis in developed countries, with an estimated incidence of 10-12 cases per million person-years. [3] The disease predominantly affects individuals in their sixth and seventh decades, with a slight male predominance. Unlike multiple myeloma, the plasma cell burden in AL amyloidosis is typically low (usually less than 10% bone marrow plasma cells), but the produced light chains have unique biochemical properties that promote misfolding and tissue deposition. [4]
The clinical significance of AL amyloidosis lies in its protean manifestations and poor prognosis when cardiac involvement is present. Early diagnosis and treatment targeting the underlying clonal plasma cell population can halt amyloid deposition and potentially reverse organ damage, making prompt recognition critical. [5] The median survival varies dramatically based on the extent and severity of cardiac involvement, ranging from less than 6 months in advanced cardiac disease (Mayo Stage IV) to several years in limited disease. [6]
2. Epidemiology
AL amyloidosis is a rare disease with distinct epidemiological characteristics that are important for clinical recognition and screening strategies.
Incidence and Prevalence
| Statistic | Value | Source |
|---|---|---|
| Annual incidence (USA/UK) | 10-12 per million | [3] |
| Estimated prevalence | 30-40 per million | [3] |
| Male:Female ratio | 1.3-2:1 | [7] |
| Median age at diagnosis | 63-65 years | [7] |
| Age range | Rarely less than 40 years; peaks 60-70 years | [7] |
| Lambda:Kappa ratio | 3:1 (75% lambda, 25% kappa) | [8] |
Demographic Patterns
The disease shows a slight male predominance and is exceptionally rare in individuals under 40 years of age. [7] The median age at diagnosis is 63-65 years, approximately 10 years younger than patients with wild-type ATTR amyloidosis, an important differential diagnosis. [9] There is no clear ethnic predisposition, though some studies suggest slightly higher rates in Caucasian populations compared to African or Asian populations. [3]
Risk Factors
While AL amyloidosis can arise de novo, several conditions are associated with increased risk:
- Monoclonal gammopathy of undetermined significance (MGUS): Approximately 10% of patients with MGUS may progress to AL amyloidosis or related disorders over time. [10]
- Smoldering multiple myeloma: Rare progression to AL amyloidosis can occur.
- Pre-existing plasma cell dyscrasias: Low-burden clonal disorders may evolve into amyloidogenic clones.
Temporal Trends
Incidence appears stable over recent decades, though improved diagnostic techniques and increased awareness have led to earlier detection. [3] The development of novel therapies, particularly proteasome inhibitors and monoclonal antibodies, has significantly improved outcomes over the past 15 years. [11]
3. Aetiology & Pathophysiology
Primary Aetiology
AL amyloidosis results from a clonal population of plasma cells or B lymphocytes in the bone marrow that produces abnormal monoclonal immunoglobulin light chains. [1] The plasma cell burden is typically small (median 5-10% of bone marrow cellularity), far lower than in symptomatic multiple myeloma (> 10%, typically > 30%). [4] In approximately 75% of cases, the abnormal light chain is of lambda type; in 25% it is kappa. [8]
Molecular Pathophysiology
Light Chain Misfolding and Fibrillogenesis
The pathogenesis of AL amyloidosis involves a multi-step process:
-
Clonal Light Chain Production: A clonal plasma cell population produces immunoglobulin light chains with specific amino acid sequence variations, particularly in the variable domain (VL). [2] Certain germline genes (particularly IGΛV6-57 and IGLV1-44 for lambda light chains) are overrepresented in AL amyloidosis. [12]
-
Thermodynamic Instability: The produced light chains have reduced thermodynamic stability and increased propensity to unfold from their native structure. Specific amino acid substitutions in the VL domain reduce the stability of the immunoglobulin fold. [13]
-
Misfolding and Aggregation: Under physiological conditions, these unstable light chains partially unfold and expose hydrophobic regions normally buried in the protein core. This triggers aggregation into oligomeric intermediates. [2]
-
β-Pleated Sheet Formation: The oligomers undergo conformational rearrangement into highly ordered, antiparallel β-pleated sheet structures. These β-sheets are the hallmark of all amyloid fibrils and confer resistance to proteolysis. [14]
-
Fibril Formation: The β-sheets stack perpendicular to the fibril axis, forming rigid, non-branching fibrils 7-10 nm in diameter. Electron microscopy reveals these characteristic ultrastructural features. [14]
-
Tissue Deposition: Amyloid fibrils deposit in the extracellular space of various organs. The pattern of organ involvement may relate to specific light chain sequence characteristics, though mechanisms determining organ tropism remain incompletely understood. [15]
Congo Red Staining and Birefringence
Amyloid fibrils have a unique affinity for Congo red dye, which binds to the β-pleated sheet structure. Under polarized light microscopy, Congo red-stained amyloid exhibits characteristic apple-green birefringence, the pathological gold standard for amyloid diagnosis. [16] This optical property results from the ordered alignment of dye molecules along the fibril axis.
Organ-Specific Pathophysiology
Cardiac Involvement
Amyloid fibrils deposit in the myocardial interstitium, leading to:
- Progressive ventricular wall thickening without true hypertrophy (increased wall thickness with normal or reduced myocyte size)
- Restrictive physiology with impaired ventricular filling and diastolic dysfunction
- Reduction in myocardial contractility despite preserved ejection fraction in early stages
- Atrial involvement with increased risk of atrial fibrillation and atrial thrombus
- Conduction system infiltration causing bradyarrhythmias and heart block
- Direct myocyte toxicity from circulating light chains, independent of fibril deposition [17]
Renal Involvement
Glomerular deposition of amyloid fibrils in the mesangium and along capillary loops causes:
- Increased glomerular permeability leading to nephrotic-range proteinuria (> 3.5 g/24h)
- Progressive glomerulosclerosis and declining GFR
- Tubular atrophy and interstitial fibrosis in advanced disease
- Vascular involvement may contribute to hypertension in some patients [18]
Hepatic Involvement
Amyloid deposits in hepatic sinusoids and vessel walls lead to:
- Hepatomegaly, often massive (palpable > 5 cm below costal margin)
- Cholestatic pattern of liver enzyme elevation (elevated alkaline phosphatase disproportionate to transaminases)
- Rarely, portal hypertension and hepatic synthetic dysfunction [18]
Peripheral and Autonomic Nervous System
- Amyloid deposits in peripheral nerve endoneurium and perineurium cause axonal degeneration
- Typically presents as distal symmetric sensorimotor polyneuropathy
- Autonomic involvement affects cardiovascular reflexes, gastrointestinal motility, and erectile function
- Small fiber neuropathy may cause pain and temperature sensation loss [19]
Soft Tissue Manifestations
- Macroglossia: Amyloid infiltration of tongue (pathognomonic when present, seen in ~10% of patients)
- Periorbital purpura: Amyloid infiltration of dermal capillaries causing fragility and spontaneous purpura, often precipitated by minor trauma or Valsalva maneuvers
- Carpal tunnel syndrome: Median nerve compression from amyloid deposition in flexor retinaculum [20]
4. Clinical Presentation
The clinical presentation of AL amyloidosis is highly variable and depends on the pattern and extent of organ involvement. Most patients present with symptoms referable to one or two organ systems, though multi-organ involvement is typically present by the time of diagnosis.
Cardinal Symptoms
Cardiac Symptoms
- Progressive dyspnea on exertion (most common presenting symptom in cardiac AL)
- Orthopnea and paroxysmal nocturnal dyspnea
- Peripheral edema, often disproportionate to severity of dyspnea
- Fatigue and reduced exercise tolerance
- Syncope or presyncope (suggesting conduction disease or autonomic dysfunction)
Renal Symptoms
- Peripheral edema (from nephrotic syndrome)
- Frothy urine (proteinuria)
- Fatigue (from anemia and hypoalbuminemia)
Neurological Symptoms
- Paresthesias and numbness in distal extremities
- Autonomic symptoms: orthostatic hypotension, early satiety, alternating diarrhea/constipation, erectile dysfunction
- Carpal tunnel syndrome (may precede systemic diagnosis by months to years)
Constitutional Symptoms
- Unintentional weight loss (often dramatic, > 10 kg)
- Fatigue and weakness (multifactorial: cardiac, anemia, autonomic)
- Anorexia
Physical Examination Findings
| System | Findings | Frequency | Clinical Significance |
|---|---|---|---|
| Cardiovascular | Elevated JVP | Common | Restrictive filling |
| Peripheral edema | 60-70% | Cardiac or renal | |
| S3 gallop | Common | Restrictive physiology | |
| Hypotension | 30-40% | Autonomic or cardiac | |
| Atrial fibrillation | 10-20% | Atrial involvement | |
| Hepatic | Hepatomegaly | 25-35% | Hepatic infiltration |
| Non-tender, firm liver | Common | Amyloid deposition | |
| Dermatological | Periorbital purpura | 15-20% | Highly specific |
| Purpura at flexural sites | Occasional | Amyloid angiopathy | |
| Oropharyngeal | Macroglossia | 10-15% | Pathognomonic |
| Dental indentations on tongue | Occasional | Tongue enlargement | |
| Neurological | Distal sensory loss | 20-30% | Peripheral neuropathy |
| Postural hypotension (> 20 mmHg drop) | 15-25% | Autonomic neuropathy | |
| Thenar atrophy | Occasional | Carpal tunnel | |
| Musculoskeletal | Shoulder pad sign | Rare | Soft tissue deposition |
| Muscular pseudohypertrophy | Rare | Muscle infiltration |
Patterns of Organ Involvement
Studies of large AL amyloidosis cohorts demonstrate the following organ involvement frequencies at diagnosis: [21]
- Kidney: 50-60%
- Heart: 50-60%
- Liver: 15-25%
- Peripheral nervous system: 15-20%
- Autonomic nervous system: 15-20%
- Gastrointestinal tract: 10-15%
- Soft tissues: 15-20%
Approximately 70% of patients have two or more organs involved at diagnosis. [21] The specific combination of organ involvement has prognostic implications, with cardiac involvement being the dominant determinant of survival.
Sentinel Clinical Presentations
Classic Presentation Patterns
-
Cardiac-Dominant Presentation: Middle-aged male with progressive dyspnea, peripheral edema, and low-voltage ECG despite increased left ventricular wall thickness on echocardiography. This discordance (low voltage with thick walls) is highly suggestive of cardiac amyloidosis.
-
Renal-Dominant Presentation: Nephrotic syndrome with massive proteinuria (often > 10 g/24h), hypoalbuminemia, and peripheral edema in the absence of diabetes or other glomerular diseases. Kidney size typically normal or enlarged (unlike chronic kidney disease).
-
Multi-System Presentation: Combination of unexplained heart failure, proteinuria, peripheral neuropathy, and hepatomegaly in an older adult. This constellation should always prompt evaluation for systemic amyloidosis.
-
Soft Tissue Presentation: Carpal tunnel syndrome (especially bilateral), periorbital purpura, or macroglossia with or without systemic symptoms. These findings are highly specific and warrant amyloid evaluation.
Red Flag Features
Several clinical features should prompt urgent evaluation and treatment:
- Rapidly progressive heart failure symptoms
- Syncope or presyncope (risk of sudden cardiac death)
- Severe orthostatic hypotension (systolic drop > 30 mmHg)
- Worsening renal function with nephrotic-range proteinuria
- Significant weight loss (> 10% body weight)
- Evidence of multi-organ system involvement
5. Differential Diagnosis
The protean manifestations of AL amyloidosis require consideration of multiple differential diagnoses depending on the presenting organ system. However, the most critical differentials are other forms of systemic amyloidosis, as these have different etiologies, treatments, and prognoses.
Primary Differentials: Other Amyloidoses
| Type | Protein | Key Distinguishing Features | Investigation Findings | Treatment |
|---|---|---|---|---|
| AL | Light chain (λ or κ) | Clonal plasma cell disorder; lambda > kappa; multi-organ; rapid progression | Abnormal sFLC ratio; M-protein; mass spec confirms AL | Chemotherapy targeting plasma cells |
| ATTR (Hereditary) | Mutant transthyretin | Family history; endemic in certain populations (V30M, V122I); younger age; neuropathy-dominant variants | Genetic testing positive; normal sFLC; mass spec confirms ATTR | Liver transplant (selected); tafamidis; patisiran |
| ATTR (Wild-type) | Wild-type transthyretin | Elderly males (> 70 yrs); isolated cardiac involvement; slow progression | Normal sFLC; no TTR mutation; mass spec confirms ATTR; normal SAP scan | Supportive; tafamidis |
| AA | Serum amyloid A | Chronic inflammatory disease (RA, IBD, chronic infection); kidney-dominant | Elevated SAA; inflammatory markers elevated; mass spec confirms AA | Treat underlying inflammation; anti-IL-1 therapy |
| Aβ2M | β2-microglobulin | Long-term dialysis (> 10 years); carpal tunnel; bone cysts; arthropathy | History of dialysis; elevated β2M; mass spec confirms Aβ2M | Kidney transplant; improved dialysis |
Critical Differentiation: AL vs. ATTR
This distinction is crucial as treatments differ completely:
Favoring AL:
- Age less than 70 years (though can occur at any age > 40)
- Multi-organ involvement (kidney, liver, neuropathy in addition to heart)
- Abnormal serum free light chain ratio
- Presence of M-protein on immunofixation
- Lambda light chain predominance (3:1)
- Rapid symptom progression (months)
- Macroglossia or periorbital purpura
Favoring ATTR:
- Age > 70 years (wild-type) or family history (hereditary)
- Isolated or dominant cardiac involvement
- Normal serum free light chain ratio
- No M-protein
- Prior carpal tunnel surgery (often years before cardiac symptoms)
- Slow progression (years)
- Biceps tendon rupture or lumbar spinal stenosis
Favoring AA:
- Long-standing chronic inflammatory condition (rheumatoid arthritis, inflammatory bowel disease, chronic osteomyelitis, familial Mediterranean fever)
- Dominant kidney involvement (nephrotic syndrome) with less cardiac involvement
- Normal serum free light chains
- Elevated inflammatory markers (CRP, ESR)
- Elevated serum amyloid A protein
Secondary Differentials by Presentation
When Presenting as Heart Failure:
- Hypertensive heart disease (but LV wall thickness proportionate to BP history)
- Hypertrophic cardiomyopathy (asymmetric hypertrophy; family history; dynamic LVOT obstruction)
- Ischemic cardiomyopathy (regional wall motion abnormalities; CAD on angiography)
- Restrictive cardiomyopathies (sarcoidosis, hemochromatosis, endomyocardial fibrosis)
- Athlete's heart (symmetric LVH; history of intense training; normal diastolic function)
When Presenting as Nephrotic Syndrome:
- Diabetic nephropathy (diabetes history; diabetic retinopathy)
- Focal segmental glomerulosclerosis (FSGS)
- Membranous nephropathy (idiopathic or secondary)
- Minimal change disease (less common in adults)
- Light chain deposition disease (LCDD - different from AL; linear deposits on immunofluorescence)
When Presenting as Peripheral Neuropathy:
- Diabetic neuropathy
- Chronic inflammatory demyelinating polyneuropathy (CIDP)
- Paraneoplastic neuropathy
- Hereditary neuropathies (Charcot-Marie-Tooth disease)
- Vitamin B12 deficiency
- Monoclonal gammopathy-associated neuropathy (MGUS with neuropathy, but no amyloid)
6. Investigations
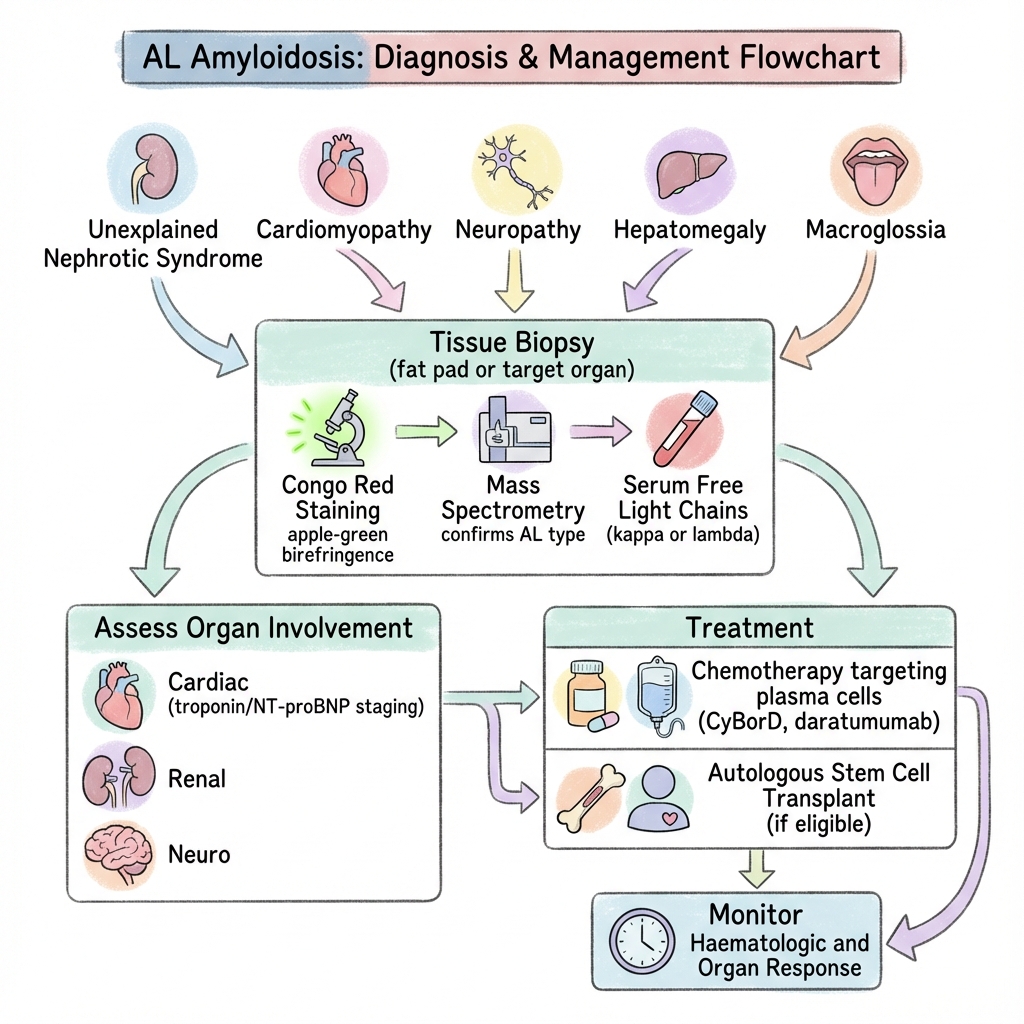
The diagnosis of AL amyloidosis requires three essential components: [22]
- Demonstration of amyloid deposits (Congo red staining with apple-green birefringence)
- Typing of the amyloid fibril protein as light chain-derived (by mass spectrometry or immunohistochemistry)
- Evidence of a clonal plasma cell or lymphocyte population (abnormal serum free light chains, M-protein, or bone marrow clonal plasma cells)
First-Line Investigations
Screening for Monoclonal Protein
| Investigation | Findings in AL Amyloidosis | Sensitivity | Notes |
|---|---|---|---|
| Serum free light chains (sFLC) | Abnormal κ/λ ratio (less than 0.26 or > 1.65) | > 95% | Most sensitive screening test; elevated involved FLC |
| Serum protein electrophoresis (SPEP) | M-protein spike (usually small less than 1 g/dL) | 50-70% | Less sensitive than sFLC; may be absent |
| Serum immunofixation | Monoclonal band (IgG, IgA, or light chain only) | 70-90% | More sensitive than SPEP |
| Urine protein electrophoresis (UPEP) | Bence Jones protein (free light chains) | 60-80% | 24-hour urine collection |
| Urine immunofixation | Monoclonal light chain | 70-90% | Complements serum testing |
The combination of serum free light chains, serum immunofixation, and urine immunofixation detects a monoclonal protein in > 99% of AL amyloidosis cases. [8] A normal serum free light chain ratio makes AL amyloidosis highly unlikely.
Tissue Biopsy for Amyloid Detection
| Biopsy Site | Sensitivity | Advantages | Disadvantages |
|---|---|---|---|
| Abdominal fat pad aspirate | 75-85% | Non-invasive; bedside procedure; safe | Lower sensitivity; may miss organ-limited disease |
| Bone marrow biopsy | 50-60% | Simultaneously assesses plasma cell burden | Lower sensitivity for amyloid detection |
| Rectal biopsy | 70-80% | Good sensitivity | More invasive; requires endoscopy |
| Kidney biopsy | > 95% (if kidney involved) | Highest sensitivity in renal disease; provides prognostic information | Invasive; bleeding risk |
| Cardiac biopsy (endomyocardial) | > 95% (if heart involved) | Definitive for cardiac amyloid | High risk; rarely needed with non-invasive imaging |
| Labial salivary gland | 60-75% | Accessible | Requires minor surgical procedure |
Recommended Diagnostic Approach:
- Start with abdominal fat pad aspirate (safest, easiest)
- If negative but clinical suspicion high, proceed to organ biopsy (kidney if renal involvement, cardiac if isolated cardiac symptoms)
- Bone marrow biopsy should be performed regardless (to assess plasma cell burden), but is inadequate alone for amyloid detection
Histological Confirmation
- Congo red staining: Amyloid appears pink-red under bright-field microscopy
- Polarized light microscopy: Characteristic apple-green birefringence (pathognomonic) [16]
- Electron microscopy: Non-branching fibrils 7-10 nm diameter (not required for diagnosis)
Amyloid Typing (Mandatory)
Once amyloid is detected, typing is essential to distinguish AL from other amyloidoses:
- Laser microdissection and mass spectrometry: Gold standard; > 95% accurate; distinguishes AL from ATTR, AA, and other types [23]
- Immunohistochemistry: Uses antibodies against kappa, lambda, TTR, SAA, etc. Less reliable; false positives/negatives occur
- Immunofluorescence: Can identify light chain restriction (kappa vs. lambda)
Mass spectrometry is now considered mandatory for definitive typing, as immunohistochemistry can give misleading results. [23]
Organ Involvement Assessment
Cardiac Investigations
| Test | Findings | Diagnostic/Prognostic Value |
|---|---|---|
| ECG | Low voltage in limb leads; pseudoinfarct pattern (QS waves); conduction abnormalities | Low voltage despite echo-LVH highly suggestive |
| Echocardiography | Increased LV/RV wall thickness; diastolic dysfunction; restrictive filling; reduced longitudinal strain; biatrial enlargement; pericardial effusion; "sparkling" granular texture | Key diagnostic and prognostic tool |
| Cardiac MRI | Diffuse subendocardial late gadolinium enhancement (LGE); abnormal gadolinium kinetics (difficulty nulling myocardium); elevated native T1; elevated ECV | > 90% sensitivity; LGE pattern highly specific |
| NT-proBNP | Markedly elevated (often > 1000 pg/mL) | Staging and prognosis (Mayo criteria) |
| Cardiac troponin T or I | Elevated (troponin T > 0.06 ng/mL) | Staging and prognosis (Mayo criteria) |
| 99mTc-PYP or DPD scintigraphy | Absent or minimal cardiac uptake in AL (Grade 0-1); intense uptake (Grade 2-3) suggests ATTR | Differentiates AL from ATTR cardiac amyloidosis |
Cardiac Biomarkers and Mayo Staging
The Mayo Clinic developed and validated staging systems for AL amyloidosis based on cardiac biomarkers:
Mayo 2004 Staging System: [24]
- Based on troponin T (cTnT ≥0.035 ng/mL) and NT-proBNP (≥332 ng/L)
- Stage I: Both normal
- Stage II: One abnormal
- Stage III: Both abnormal
- Median survival: Stage I (26.4 months), Stage II (10.5 months), Stage III (3.5 months)
Mayo 2012 Staging System (Revised): [6] Adds difference in free light chains (dFLC):
- Biomarkers: NT-proBNP > 1800 ng/L (1 point), cTnT > 0.025 ng/mL (1 point), dFLC > 180 mg/L (1 point)
- Stage I: 0 points (median survival 94 months)
- Stage II: 1 point (median survival 40 months)
- Stage III: 2 points (median survival 14 months)
- Stage IV: 3 points (median survival 6 months)
This staging system is the most widely used prognostic tool and guides treatment decisions. [6]
Echocardiographic Findings
Key features suggesting cardiac amyloidosis:
- Increased LV wall thickness (typically concentric) with preserved or reduced ejection fraction
- Normal or small LV cavity size
- Biatrial enlargement (amyloid involves atria as well)
- Thickened interatrial septum
- Thickened right ventricular free wall
- Small pericardial effusion (common)
- Restrictive filling pattern on Doppler (E/A > 2, short deceleration time)
- Reduced global longitudinal strain (GLS) with apical sparing ("cherry on top" pattern)
- Granular, "sparkling" myocardial texture (subjective, not specific)
Cardiac MRI in AL Amyloidosis
Cardiac MRI features highly suggestive of amyloidosis:
- Diffuse, global subendocardial or transmural late gadolinium enhancement (unlike ischemic cardiomyopathy, which shows subendocardial/transmural LGE in coronary territories)
- Difficulty "nulling" the myocardium on inversion recovery sequences (amyloid fibrils alter gadolinium kinetics)
- Elevated native T1 values (> 1100 ms at 1.5T)
- Elevated extracellular volume (ECV > 40%)
- LGE of atria and right ventricle (indicates advanced disease)
Renal Investigations
- 24-hour urine protein: Typically > 3.5 g/24h (nephrotic range); may exceed 10 g/24h
- Serum albumin: Low (less than 3.5 g/dL) due to urinary losses
- Serum creatinine/eGFR: Variable; often preserved until late disease
- Kidney size on ultrasound: Normal or enlarged (unlike most chronic kidney diseases)
- Kidney biopsy: Gold standard for renal amyloid; shows mesangial and glomerular capillary loop deposits
Hepatic Investigations
- Liver function tests: Elevated alkaline phosphatase (ALP) disproportionate to transaminases (cholestatic pattern)
- Hepatomegaly on imaging: Ultrasound or CT shows enlarged, homogeneous liver
- Liver biopsy: Rarely needed; shows amyloid in sinusoids and vessel walls
Neurological Investigations
- Nerve conduction studies/EMG: Axonal sensorimotor polyneuropathy (reduced amplitudes)
- Autonomic function testing: Abnormal heart rate variability; orthostatic BP changes
- Quantitative sudomotor axon reflex test (QSART): Assesses small fiber/autonomic function
Staging Investigations
Bone Marrow Examination
- Bone marrow aspirate and biopsy: Assess plasma cell percentage (usually less than 10% in AL amyloidosis vs. > 10% in myeloma)
- Flow cytometry: Identifies clonal plasma cells; immunophenotyping
- Cytogenetics and FISH: Assess for high-risk cytogenetic abnormalities (t(11;14), gain 1q21, del 17p); prognostic implications [25]
Whole-Body Imaging
- Serum amyloid P (SAP) scintigraphy: Radiolabeled SAP localizes to amyloid deposits; assesses extent and distribution (available in limited centers; excellent for quantifying visceral amyloid burden) [26]
- PET/CT: Not standard but may show organ involvement
Differential Free Light Chain (dFLC) Measurement
The dFLC is calculated as the absolute difference between the involved and uninvolved free light chains. [6]
- dFLC = |involved FLC - uninvolved FLC|
- High dFLC (> 180 mg/L) indicates high clonal light chain burden and is associated with poorer prognosis (Mayo 2012 staging)
- dFLC is also used to monitor treatment response (see Management section)
7. Classification and Staging
Amyloidosis Classification
AL amyloidosis is classified as one form of systemic amyloidosis:
Systemic Amyloidoses:
- AL (Light chain): Clonal plasma cell disorder → immunoglobulin light chains
- AA (Inflammatory): Chronic inflammation → serum amyloid A protein
- ATTR (Transthyretin):
- Wild-type (senile cardiac amyloidosis)
- Hereditary (mutant TTR; > 120 mutations identified)
- Aβ2M (Dialysis-related): Long-term dialysis → β2-microglobulin
- Others: Rare (fibrinogen, apolipoprotein, lysozyme, etc.)
Localized Amyloidoses:
- Do not involve systemic organs; often AL type localized to single site (bladder, skin, airways)
Mayo Clinic Staging System (2012)
The most widely used prognostic staging system: [6]
| Stage | NT-proBNP | Troponin T | dFLC | Points | Median OS |
|---|---|---|---|---|---|
| I | less than 1800 ng/L | less than 0.025 ng/mL | less than 180 mg/L | 0 | 94 months |
| II | One abnormal | 1 | 40 months | ||
| III | Two abnormal | 2 | 14 months | ||
| IV | All three abnormal | 3 | 6 months |
- NT-proBNP > 1800 ng/L: 1 point
- Troponin T > 0.025 ng/mL (or troponin I > 0.1 ng/mL): 1 point
- dFLC > 180 mg/L: 1 point
This system stratifies patients into four risk groups with significantly different median overall survival, and guides treatment intensity and transplant eligibility.
Renal Staging
Renal involvement can be staged based on proteinuria and GFR:
- Stage 1: Proteinuria > 0.5 g/24h; eGFR > 50 mL/min
- Stage 2: Proteinuria > 0.5 g/24h; eGFR less than 50 mL/min
- Stage 3: Requiring dialysis
Renal stage influences treatment choice and eligibility for autologous stem cell transplantation.
European Staging System
Alternative system using only NT-proBNP and troponin (without dFLC), similar to Mayo 2004 criteria. Less commonly used now.
8. Management
The management of AL amyloidosis has two primary goals: [11]
- Eradicate or suppress the clonal plasma cell population producing amyloidogenic light chains (prevents further amyloid deposition and allows existing deposits to gradually regress)
- Provide organ-specific supportive care to manage complications of organ dysfunction
Treatment decisions are guided by Mayo stage, organ involvement, performance status, and patient factors (age, comorbidities).
Treatment Algorithm Overview
AL Amyloidosis Diagnosis Confirmed
↓
Assess Eligibility
↓
┌──────┴──────┐
↓ ↓
ASCT Eligible ASCT Ineligible
(Mayo I-II, (Mayo III-IV, age > 70,
age less than 70, severe cardiac involvement,
adequate poor performance status)
organ function)
↓ ↓
│ Chemotherapy:
│ • D-CyBorD (preferred)
│ • CyBorD
│ • Bortezomib-dexamethasone
↓
Induction chemotherapy
(CyBorD or Bor-Dex)
↓
Stem cell mobilization
↓
High-dose melphalan + ASCT
↓
Consolidation if needed
↓
Monitor for response/relapse
First-Line Chemotherapy
Daratumumab-Based Regimens (Current Standard)
The ANDROMEDA trial (2021) established subcutaneous daratumumab combined with CyBorD (D-CyBorD) as the new standard of care for newly diagnosed AL amyloidosis. [27]
ANDROMEDA Trial Results:
- Design: Phase 3 RCT comparing D-CyBorD vs. CyBorD alone
- Primary endpoint: Hematologic complete response (CR) rate at 6 months
- Results:
- "Hematologic CR: 53% (D-CyBorD) vs. 18% (CyBorD alone); pless than 0.001"
- "Major organ deterioration or death: 36.6% (D-CyBorD) vs. 44.8% (CyBorD)"
- "Cardiac response rate: significantly higher with daratumumab"
- Conclusion: D-CyBorD is superior to CyBorD and is now considered standard first-line therapy [27]
D-CyBorD Regimen:
- Daratumumab: 16 mg/kg subcutaneous weekly (cycles 1-2), every 2 weeks (cycles 3-6), then every 4 weeks
- Cyclophosphamide: 300 mg/m² PO weekly
- Bortezomib: 1.3 mg/m² subcutaneous weekly (days 1, 8, 15, 22 of 28-day cycle)
- Dexamethasone: 20-40 mg PO weekly (reduced dose in cardiac involvement: 20 mg)
CyBorD Regimen (if daratumumab unavailable):
- Same as above but without daratumumab
- Historically, the standard of care prior to ANDROMEDA trial
- Hematologic response rates: Overall response ~60%, CR ~30% [28]
Alternative Regimens:
-
Bortezomib-Dexamethasone (Bor-Dex)
- Bortezomib 1.3 mg/m² SC twice weekly + dexamethasone 20-40 mg weekly
- Used when cyclophosphamide contraindicated
- Response rates: overall ~70%, CR ~30% [29]
-
Melphalan-Dexamethasone (Mel-Dex)
- Oral melphalan 0.22 mg/kg days 1-4 + dexamethasone 40 mg days 1-4 every 28 days
- Older regimen, less commonly used now; inferior to bortezomib-based regimens
- Avoid in transplant-eligible patients (may impair stem cell mobilization)
-
Bendamustine-based regimens
- Bendamustine + dexamethasone
- Limited evidence in AL amyloidosis; used in selected refractory cases
Dose Modifications:
- Cardiac involvement (NT-proBNP > 8500 ng/L or Mayo Stage IV): Reduce dexamethasone to 20 mg weekly (lower doses to minimize fluid retention)
- Renal impairment (eGFR less than 30): Reduce cyclophosphamide dose or avoid; adjust bortezomib dosing
- Neuropathy: Switch bortezomib to subcutaneous (reduces neuropathy risk vs. IV); consider dose reduction or alternative regimen
Autologous Stem Cell Transplantation (ASCT)
High-dose melphalan (100-200 mg/m²) followed by autologous stem cell rescue remains an effective treatment for selected patients with AL amyloidosis. [30]
Eligibility Criteria:
- Age typically ≤70 years (though older patients considered in excellent health)
- Mayo Stage I or II (Stage III-IV have prohibitively high transplant-related mortality)
- Adequate organ function:
- "Cardiac: NT-proBNP less than 5000 ng/L, troponin normal or minimally elevated, NYHA class I-II"
- "Renal: eGFR > 30 mL/min (preferably > 50)"
- ≤2 organs involved
- No severe autonomic neuropathy or uncontrolled heart failure
Transplant Outcomes:
- Hematologic CR rate: 40-50%
- Treatment-related mortality: 3-5% (in selected patients); higher in older series
- Median overall survival: 6-10 years in responding patients [30]
- Improved survival compared to conventional chemotherapy in appropriately selected patients
Transplant Procedure:
- Induction chemotherapy: 1-2 cycles of CyBorD or Bor-Dex to reduce light chain burden
- Stem cell mobilization: G-CSF with or without plerixafor
- Conditioning: High-dose melphalan 100-200 mg/m² (dose reduced in organ dysfunction)
- Stem cell reinfusion: Day 0
- Consolidation/maintenance: Not standard; under investigation
Comparison: ASCT vs. Chemotherapy Alone
While no randomized trials directly compare modern chemotherapy (D-CyBorD) to ASCT, historical data and registry studies suggest:
- ASCT achieves higher CR rates but has upfront toxicity
- Transplant-eligible patients treated with ASCT may have longer survival if they achieve hematologic CR
- Modern chemotherapy (especially D-CyBorD) has narrowed the survival gap and is preferred initial therapy in some centers, reserving ASCT for relapse
Current practice varies: some centers use D-CyBorD as first-line and ASCT at relapse; others proceed to ASCT after induction in highly selected patients.
Treatment Response Criteria
Hematologic response and organ response are assessed separately. [31]
Hematologic Response (based on serum free light chains):
- Complete response (CR): Normal FLC ratio (0.26-1.65) and negative serum/urine immunofixation
- Very good partial response (VGPR): dFLC less than 40 mg/L
- Partial response (PR): dFLC decrease ≥50%
- No response: Less than PR
- Progression: 50% increase in dFLC to > 100 mg/L
Organ Response (assessed at 3-6 months after hematologic response):
Cardiac response:
- NT-proBNP decrease > 30% and > 300 ng/L (if baseline > 650 ng/L)
- Improvement in NYHA class
Renal response:
- 24-hour proteinuria decrease ≥30% (if baseline ≥0.5 g) without ≥25% worsening of eGFR
- Complete renal response: proteinuria less than 0.5 g/24h
Hepatic response:
- Liver size reduction ≥2 cm or alkaline phosphatase decrease > 50%
Hematologic response typically precedes organ response by 3-6 months. Achieving CR or VGPR is associated with significantly improved survival and organ response rates.
Relapsed/Refractory Disease
Patients who relapse after initial therapy or do not respond require second-line treatment.
Second-Line Options:
- If initial therapy was CyBorD: Add daratumumab (D-CyBorD)
- If initial therapy included daratumumab: Consider alternative regimen (ixazomib, pomalidomide, venetoclax if t(11;14)+)
- Venetoclax: Highly active in patients with t(11;14) translocation (found in ~50% of AL amyloidosis); response rates > 50% in t(11;14)+ patients [32]
- Ixazomib-based regimens: Oral proteasome inhibitor; alternative to bortezomib
- Pomalidomide-dexamethasone: Immunomodulatory drug; effective in refractory cases
- ASCT: If not done previously and patient now eligible
Organ-Specific Supportive Management
Cardiac Management
AL cardiac amyloidosis requires careful management as standard heart failure therapies may be poorly tolerated:
-
Diuretics: Mainstay of symptom management; loop diuretics (furosemide) for volume overload
- Patients are highly preload-dependent; overly aggressive diuresis can cause hypotension and renal dysfunction
- "Goal: euvolemia without hypotension"
-
ACE inhibitors/ARBs: Generally poorly tolerated due to risk of hypotension; avoid unless hypertensive
-
Beta-blockers: May worsen symptoms due to fixed stroke volume and bradycardia; use cautiously
-
Digoxin: Contraindicated (binds to amyloid fibrils; risk of toxicity even at therapeutic levels) [33]
-
Calcium channel blockers: Contraindicated (negative inotropic effects; can bind amyloid)
-
Anticoagulation:
- "Atrial fibrillation: High risk of atrial thrombus even without AF; consider anticoagulation if AF present or documented atrial standstill"
- CHA₂DS₂-VASc score underestimates stroke risk in cardiac amyloidosis
-
Pacemaker/ICD:
- Pacemaker for symptomatic bradycardia or high-grade AV block (common in advanced disease)
- ICD generally not recommended (sudden death often from electromechanical dissociation, not VT/VF; poor prognosis limits benefit)
-
Mechanical circulatory support/transplant:
- Heart transplant contraindicated in AL amyloidosis (systemic disease will involve transplanted heart)
- May consider combined heart-kidney transplant in selected patients with isolated organ-limited amyloidosis after achieving hematologic CR
Renal Management
- Diuretics: For edema from nephrotic syndrome
- ACE inhibitors/ARBs: May reduce proteinuria but limited benefit; avoid if hypotensive
- Avoid nephrotoxins: NSAIDs, aminoglycosides, contrast (use cautiously)
- Dialysis: When eGFR declines to ESRD levels; peritoneal or hemodialysis both feasible
- Kidney transplant: Consider in patients achieving hematologic CR with sustained response (> 1 year); improved outcomes compared to dialysis [34]
Autonomic Neuropathy Management
-
Orthostatic hypotension:
- "Non-pharmacologic: Compression stockings, increase salt/fluid intake, slow postural changes"
- "Pharmacologic: Midodrine (alpha-agonist, 5-10 mg TID), fludrocortisone (mineralocorticoid, 0.1-0.2 mg daily)"
-
Gastrointestinal dysmotility:
- Prokinetics (metoclopramide), antiemetics
- Dietary modifications (small frequent meals)
-
Erectile dysfunction: Phosphodiesterase inhibitors (use cautiously in cardiac disease)
Nutritional Support
- Weight loss common; nutritional supplementation important
- Macroglossia can impair eating; soft diet may be needed
Novel and Emerging Therapies
- CAEL-101: Monoclonal antibody targeting misfolded light chains; facilitates amyloid clearance; Phase 2/3 trials ongoing [35]
- Birtamimab: Anti-amyloid fibril antibody; targets deposited amyloid
- NEOD001: Previously investigated anti-amyloid antibody (trials halted due to lack of efficacy)
- Gene silencing for ATTR (not applicable to AL): Patisiran, inotersen, vutrisiran
Treatment Monitoring
Frequency:
- Hematologic response: Assess sFLC and M-protein every 1-3 months during treatment
- Cardiac biomarkers (NT-proBNP, troponin): Every 3 months or more frequently if clinically indicated
- Organ function (renal, hepatic): Every 3 months
- Imaging (echocardiography): Every 6-12 months or if clinical change
Long-term Follow-up:
- Patients achieving CR: Monitor indefinitely for relapse (sFLC every 3-6 months)
- Median time to relapse after CR: 3-5 years; some patients remain in durable remission
9. Complications
Disease-Related Complications
| Complication | Frequency | Pathophysiology | Prevention | Management |
|---|---|---|---|---|
| Heart failure progression | > 60% | Progressive amyloid deposition; direct light chain toxicity | Achieve rapid hematologic response | Diuretics; manage volume carefully; consider transplant if eligible |
| Sudden cardiac death | 25-40% | Ventricular arrhythmias; electromechanical dissociation; bradyarrhythmias | Pacemaker for bradycardia; ICD limited benefit | Optimize cardiac therapy; avoid QT-prolonging drugs |
| Renal failure/ESRD | 20-30% | Progressive glomerular amyloid; GFR decline | Hematologic response; avoid nephrotoxins | Dialysis; consider transplant after sustained CR |
| Thromboembolic events | 10-15% | Atrial standstill; nephrotic syndrome (hypercoagulable); amyloid-related vascular fragility | Anticoagulation if AF or documented atrial thrombus | Anticoagulation (warfarin or DOAC) |
| Bleeding (GI, purpura) | 10-20% | Amyloid infiltration of vessels causing fragility; acquired factor X deficiency (rare) | Avoid anticoagulation if active bleeding | Transfusion support; factor replacement if deficiency; endoscopic therapy for GI bleeding |
| Autonomic dysfunction | 15-25% | Amyloid infiltration of autonomic nerves | Early recognition; supportive measures | Midodrine, fludrocortisone; compression stockings |
| Malnutrition/cachexia | 30-50% | GI dysmotility; macroglossia; hypermetabolic state | Nutritional support early | Enteral feeding if needed; small frequent meals |
Treatment-Related Complications
| Complication | Associated Treatment | Frequency | Management |
|---|---|---|---|
| Peripheral neuropathy | Bortezomib | 20-35% | Switch to subcutaneous; dose reduction; alternative agent |
| Herpes zoster reactivation | Bortezomib, daratumumab | 10-15% | Acyclovir prophylaxis (recommended) |
| Infusion reactions | Daratumumab | 40-50% (first dose) | Premedication; slow infusion; subcutaneous formulation preferred |
| Cytopenias | Chemotherapy, ASCT | Variable | Dose reduction; growth factor support; transfusion |
| Infection | Immunosuppression, ASCT | 15-30% | Antimicrobial prophylaxis; vaccinations (prior to treatment); prompt treatment |
| Transplant-related mortality | ASCT | 3-5% | Careful patient selection; experienced center |
| Fluid retention | Dexamethasone | Common | Reduce dose in cardiac patients (20 mg vs. 40 mg); diuretics |
Organ-Specific Complications
Cardiac:
- Atrial fibrillation (10-20%): High stroke risk; consider anticoagulation
- Complete heart block: May require pacemaker
- Pericardial effusion: Usually small; rarely causes tamponade
Renal:
- Nephrotic syndrome complications: Edema, VTE, infections, hyperlipidemia
- Renal vein thrombosis (rare)
Hepatic:
- Hepatic rupture (very rare; spontaneous liver rupture from massive amyloid burden)
- Portal hypertension (rare)
Gastrointestinal:
- GI bleeding from mucosal amyloid infiltration
- Intestinal pseudo-obstruction
- Malabsorption
10. Prognosis
The prognosis of AL amyloidosis has improved significantly with modern therapies, but remains highly variable depending on cardiac involvement and treatment response.
Overall Survival by Mayo Stage
| Mayo Stage | Median OS (Historical, Conventional Chemo) | Median OS (Modern Era, D-CyBorD/ASCT) |
|---|---|---|
| I | 94 months | Not reached (> 10 years) |
| II | 40 months | 5-7 years |
| III | 14 months | 2-4 years |
| IV | 6 months | 12-18 months |
Prognostic Factors
Favorable Prognostic Factors:
- Mayo Stage I-II (low cardiac biomarkers, low dFLC)
- Absence of cardiac involvement
- Achievement of hematologic complete response (CR)
- Early diagnosis (before extensive organ damage)
- Lambda light chain (slightly better prognosis than kappa)
- Low plasma cell burden
- Absence of high-risk cytogenetics (t(4;14), del 17p, gain 1q21)
Unfavorable Prognostic Factors:
- Mayo Stage III-IV (high cardiac biomarkers)
- Severe cardiac involvement (NT-proBNP > 8500 ng/L, troponin elevation)
- Multi-organ involvement (≥3 organs)
- High dFLC (> 180 mg/L)
- Failure to achieve hematologic response
- High-risk cytogenetics
- Older age (> 75 years)
- Poor performance status
Impact of Treatment Response
Achieving hematologic CR is the single most important predictor of long-term survival:
- CR achieved: Median OS > 10 years [31]
- VGPR achieved: Median OS 5-7 years
- PR or less: Median OS 2-3 years
- No response: Median OS less than 1 year
Organ response follows hematologic response by several months, but once achieved, significantly improves outcomes. Cardiac response (decrease in NT-proBNP) is associated with improved survival.
Natural History if Untreated
Without treatment, AL amyloidosis is uniformly fatal:
- Median survival (untreated, cardiac involvement): 6-12 months
- Median survival (untreated, without cardiac involvement): 2-3 years
Survival Improvements Over Time
With the introduction of novel agents (bortezomib, daratumumab) and improved supportive care, median overall survival has improved from approximately 1-2 years (1990s) to 4-5 years (2010s) and continues to improve with current therapies. [11]
Causes of Death
- Cardiac causes: 40-60% (progressive heart failure, sudden cardiac death, arrhythmias)
- Infection: 10-20% (immunosuppression from disease and treatment)
- Renal failure: 5-10%
- Other organ failure: Variable
- Treatment-related: less than 5% (mainly ASCT-related in transplant-eligible patients)
11. Prevention and Screening
Primary Prevention
There are no established primary prevention strategies for AL amyloidosis, as the disease arises sporadically from clonal plasma cell disorders.
Screening Recommendations
High-Risk Populations:
Screening for AL amyloidosis should be considered in:
-
Patients with MGUS (monoclonal gammopathy of undetermined significance) presenting with:
- Unexplained heart failure with increased wall thickness
- Nephrotic syndrome
- Peripheral neuropathy
- Hepatomegaly
-
Patients with unexplained nephrotic syndrome (check serum free light chains)
-
Patients with unexplained cardiac hypertrophy and:
- Low-voltage ECG
- Diastolic dysfunction
- Restrictive physiology
-
Patients with bilateral carpal tunnel syndrome followed by systemic symptoms
Screening Tests:
- Serum free light chains (sFLC) and kappa/lambda ratio: First-line screening test
- Serum and urine immunofixation if sFLC abnormal
- If positive, proceed to tissue biopsy (fat pad, bone marrow, organ biopsy)
MGUS Monitoring:
Patients with MGUS should be monitored for development of AL amyloidosis or other plasma cell disorders:
- Annual serum free light chains
- Annual complete blood count, creatinine, calcium
- Investigate symptoms (fatigue, edema, dyspnea, neuropathy) promptly
Genetic Counseling
AL amyloidosis is not hereditary (unlike hereditary ATTR amyloidosis). No genetic counseling or family screening is indicated.
12. Key Guidelines
International Society of Amyloidosis (ISA) Consensus
The International Society of Amyloidosis publishes periodic consensus recommendations on diagnosis and treatment of amyloidosis:
- Diagnostic criteria: Require tissue biopsy with Congo red positivity and amyloid typing by mass spectrometry or immunohistochemistry
- Organ involvement criteria: Standardized definitions for cardiac, renal, hepatic, neurological, and soft tissue involvement [21]
- Response criteria: Hematologic and organ response assessments [31]
European Society of Cardiology (ESC) Guidelines
ESC guidelines on cardiomyopathies include recommendations for cardiac amyloidosis:
- Echocardiography and cardiac MRI for diagnosis
- Cardiac biomarkers (NT-proBNP, troponin) for staging and prognosis
- Avoidance of digoxin and certain other medications
- Anticoagulation in atrial fibrillation
National Comprehensive Cancer Network (NCCN) Guidelines
NCCN provides treatment guidelines for AL amyloidosis as part of systemic light chain amyloidosis recommendations:
- First-line therapy: Daratumumab-based regimens (D-CyBorD) preferred
- ASCT in eligible patients after risk stratification
- Response-adapted therapy based on hematologic and organ response
British Society for Haematology (BSH) Guidelines
BSH guidelines on diagnosis and management of AL amyloidosis (updated periodically):
- Emphasis on rapid diagnosis and treatment initiation
- Multidisciplinary approach involving haematology, cardiology, nephrology
- Bortezomib-based chemotherapy as backbone of treatment
- Incorporation of daratumumab as standard of care
Common Examination Questions
Written Examination Questions
-
What is the pathophysiology of AL amyloidosis?
- Focus on: clonal plasma cell disorder, light chain production, misfolding, β-pleated sheet formation, Congo red staining, organ deposition
-
How is AL amyloidosis diagnosed?
- Tissue biopsy (Congo red, apple-green birefringence), amyloid typing (mass spectrometry), evidence of clonal plasma cells (serum free light chains, immunofixation, bone marrow)
-
What are the Mayo Clinic staging criteria for AL amyloidosis?
- Mayo 2012: NT-proBNP > 1800 ng/L, troponin T > 0.025 ng/mL, dFLC > 180 mg/L; stages I-IV based on 0-3 points
-
What is the first-line treatment for AL amyloidosis?
- Daratumumab-based chemotherapy (D-CyBorD); autologous stem cell transplant in eligible patients
-
How do you differentiate AL from ATTR cardiac amyloidosis?
- AL: younger age, multi-organ involvement, abnormal serum free light chains, lambda predominance
- ATTR: older age (> 70), isolated cardiac, normal free light chains, 99mTc-PYP/DPD scan positive
Clinical Examination (OSCE/PACES) Scenarios
Station: Cardiovascular Examination
Findings: Elevated JVP, peripheral edema, low-volume pulse, apex beat not displaced, added S3 gallop, hepatomegaly
Question: "What are the possible causes of this patient's heart failure?"
Expected answer: Discuss restrictive cardiomyopathy; mention AL amyloidosis, ATTR amyloidosis, sarcoidosis, hemochromatosis. Note that cardiac amyloidosis should be suspected with heart failure, increased wall thickness on echo, and low-voltage ECG.
Station: History Taking
Stem: 65-year-old man with progressive dyspnea, ankle swelling, weight loss
Key points to elicit:
- Cardiac symptoms (dyspnea, edema, orthopnea)
- Renal symptoms (frothy urine)
- Neurological symptoms (paresthesias, dizziness on standing)
- Systemic symptoms (weight loss, fatigue)
- Past history of carpal tunnel syndrome or other suggestive features
Viva Preparation
Opening Statement
"AL amyloidosis is a systemic disease caused by extracellular deposition of misfolded immunoglobulin light chains produced by a clonal plasma cell disorder. The light chains form insoluble amyloid fibrils with β-pleated sheet structure that deposit in organs including the heart, kidneys, liver, and peripheral nerves, leading to progressive organ dysfunction. Diagnosis requires tissue biopsy demonstrating Congo red-positive amyloid with apple-green birefringence, typing by mass spectrometry to confirm AL type, and evidence of a clonal plasma cell population through abnormal serum free light chains or bone marrow examination. Treatment targets the underlying plasma cell clone with chemotherapy, now standardly including daratumumab-based regimens, with autologous stem cell transplantation reserved for selected eligible patients. Prognosis is determined primarily by the extent of cardiac involvement, assessed using the Mayo staging system based on NT-proBNP, troponin, and difference in free light chains."
Key Facts to Mention
- Epidemiology: Incidence 10-12 per million per year; median age 63-65 years; lambda:kappa ratio 3:1
- Pathophysiology: Clonal plasma cells produce unstable light chains → misfolding → β-pleated sheets → tissue deposition → organ dysfunction
- Diagnosis: Tissue biopsy (Congo red apple-green birefringence) + typing (mass spec) + clonal evidence (abnormal sFLC ratio in > 95%)
- Mayo 2012 staging: NT-proBNP > 1800, troponin T > 0.025, dFLC > 180 mg/L; stages I-IV with median survival 94, 40, 14, 6 months
- Treatment: D-CyBorD (daratumumab + cyclophosphamide + bortezomib + dexamethasone) first-line based on ANDROMEDA trial; ASCT in eligible patients
- Prognosis: Achieving hematologic CR associated with median OS > 10 years; cardiac involvement is dominant prognostic factor
Model Answers to Common Questions
Q1: "How would you investigate a patient with suspected AL amyloidosis?"
"I would approach this systematically. First, I would screen for a monoclonal protein with serum free light chains, which are abnormal in over 95% of cases, along with serum and urine immunofixation to detect an M-protein. If positive, I would obtain tissue confirmation through a biopsy—starting with abdominal fat pad aspirate as it is non-invasive and 75-85% sensitive, or proceeding directly to organ biopsy if the fat pad is negative but suspicion remains high. The biopsy would be stained with Congo red to demonstrate apple-green birefringence under polarized light, confirming amyloid, and then typed using mass spectrometry, which is the gold standard for determining AL type and differentiating it from ATTR or AA amyloidosis. I would also perform a bone marrow biopsy to assess plasma cell burden and cytogenetics. To evaluate organ involvement, I would check NT-proBNP and troponin for cardiac staging, 24-hour urine protein and renal function for kidney involvement, liver function tests for hepatic involvement, and echocardiography and cardiac MRI to assess cardiac amyloid burden and guide prognosis using the Mayo staging system."
Q2: "How does AL amyloidosis differ from ATTR amyloidosis?"
"The key differences lie in the fibril protein, patient demographics, clinical presentation, and treatment. In AL amyloidosis, the amyloid is derived from immunoglobulin light chains produced by a clonal plasma cell disorder, whereas in ATTR, the amyloid comes from transthyretin protein, either wild-type or due to a genetic mutation. AL typically presents in patients aged 60-70 with multi-organ involvement including heart, kidneys, liver, and peripheral nerves, and shows an abnormal serum free light chain ratio. ATTR, particularly wild-type, presents in elderly males over 70 with isolated cardiac involvement and normal serum free light chains. Clinically, AL tends to progress more rapidly over months, while wild-type ATTR progresses over years. On imaging, 99mTc-PYP or DPD scintigraphy shows intense cardiac uptake in ATTR but minimal or absent uptake in AL. Treatment also differs fundamentally: AL requires chemotherapy targeting the plasma cell clone with agents like daratumumab and bortezomib, while ATTR may benefit from TTR stabilizers like tafamidis or gene silencing therapies like patisiran. Definitive differentiation requires amyloid typing by mass spectrometry."
Q3: "What is the ANDROMEDA trial and how has it changed practice?"
"The ANDROMEDA trial, published in 2021 in the New England Journal of Medicine, was a phase 3 randomized controlled trial comparing daratumumab plus CyBorD to CyBorD alone in newly diagnosed AL amyloidosis. Daratumumab is a CD38 monoclonal antibody that targets plasma cells. The primary endpoint was hematologic complete response at 6 months. Results showed a significant improvement with the addition of daratumumab: 53% achieved complete response versus 18% with CyBorD alone. Additionally, fewer patients in the daratumumab arm experienced major organ deterioration or death. Importantly, cardiac response rates were also higher with daratumumab. Based on these results, daratumumab-based therapy, specifically D-CyBorD, is now considered the standard of care for first-line treatment of AL amyloidosis, representing a major advance in achieving deeper and faster hematologic responses, which are critical for improving organ outcomes and survival."
Q4: "What cardiac medications should be avoided in AL cardiac amyloidosis and why?"
"Several medications commonly used in heart failure should be avoided or used with extreme caution in AL cardiac amyloidosis. Digoxin is contraindicated because it binds to amyloid fibrils, leading to toxicity even at therapeutic serum levels. Calcium channel blockers, particularly non-dihydropyridines like diltiazem and verapamil, should also be avoided as they can bind to amyloid and cause severe negative inotropic effects and hypotension. ACE inhibitors and ARBs are generally poorly tolerated due to the restrictive physiology and tendency toward hypotension, as these patients are highly preload-dependent. Beta-blockers can worsen symptoms by causing bradycardia and reducing cardiac output in patients with fixed stroke volumes. The mainstay of symptom management is careful diuresis with loop diuretics to achieve euvolemia without causing hypotension. Anticoagulation should be considered in atrial fibrillation due to high stroke risk. Pacemakers may be needed for bradyarrhythmias, but ICDs are generally not recommended as sudden death often results from electromechanial dissociation rather than ventricular arrhythmias, and the prognosis often does not justify the intervention."
Examiner Follow-Up Questions and Responses
Q: "What is the significance of the dFLC in AL amyloidosis?"
A: "The difference in free light chains, or dFLC, is calculated as the absolute difference between the involved and uninvolved free light chains. It reflects the clonal light chain burden. A high dFLC, specifically greater than 180 mg/L, is one of the three biomarkers in the Mayo 2012 staging system and is associated with poorer prognosis. It is also used to monitor treatment response: achieving a dFLC less than 40 mg/L defines a very good partial response, while normalization of the free light chain ratio defines complete response. The dFLC is more specific than total light chain levels and is a key parameter in both staging and response assessment."
Q: "Can patients with AL amyloidosis receive a heart transplant?"
A: "Heart transplantation alone is contraindicated in AL amyloidosis because it is a systemic disease, and the amyloidogenic light chains will continue to deposit in the transplanted heart, leading to recurrence. However, in highly selected patients who have achieved a sustained hematologic complete response, meaning the plasma cell clone has been eradicated and there is no ongoing light chain production, combined heart and kidney transplantation or heart transplant after prolonged remission has been performed in rare cases with reported success. The key is ensuring durable control of the underlying plasma cell disorder. More commonly, patients are treated with chemotherapy aimed at organ preservation, and in recent years, achieving deep hematologic responses with modern therapies has improved cardiac outcomes without need for transplantation."
Q: "What is the role of SAP scintigraphy?"
A: "Serum amyloid P component, or SAP, scintigraphy involves injecting radiolabeled SAP, which binds to amyloid deposits throughout the body. The scan can quantify and localize amyloid burden in visceral organs such as liver, spleen, and kidneys. It is highly specific for amyloid and can demonstrate reduction in amyloid load following successful treatment. However, it does not reliably detect cardiac amyloid, which limits its utility in AL amyloidosis where cardiac involvement is so critical. SAP scintigraphy is available in only a few specialized centers, primarily in the UK. While valuable for research and monitoring systemic amyloid burden, it is not essential for routine diagnosis or management, which rely more on cardiac biomarkers, echocardiography, and cardiac MRI for assessing cardiac involvement."
References
-
Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583-596. doi:10.1056/NEJMra023144
-
Blancas-Mejia LM, Ramirez-Alvarado M. Systemic amyloidoses. Annu Rev Biochem. 2013;82:745-774. doi:10.1146/annurev-biochem-072611-130030
-
Kyle RA, Larson DR, Kurtin PJ, et al. Incidence of AL amyloidosis in Olmsted County, Minnesota, 1990 through 2015. Mayo Clin Proc. 2019;94(3):465-471. doi:10.1016/j.mayocp.2018.08.041
-
Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am J Hematol. 2005;79(4):319-328. doi:10.1002/ajh.20381
-
Palladini G, Merlini G. What is new in diagnosis and management of light chain amyloidosis? Blood. 2016;128(2):159-168. doi:10.1182/blood-2016-01-629790
-
Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989-995. doi:10.1200/JCO.2011.38.5724
-
Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45-59.
-
Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215-224. doi:10.1038/leu.2008.307
-
Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007-1016. doi:10.1056/NEJMoa1805689
-
Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362-1369. doi:10.1056/NEJMoa054494
-
Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641-2654. doi:10.1016/S0140-6736(15)01274-X
-
Comenzo RL, Zhang Y, Martinez C, et al. The tropism of organ involvement in primary systemic amyloidosis: contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood. 2001;98(3):714-720. doi:10.1182/blood.v98.3.714
-
Baden EM, Randles EG, Aboagye AK, et al. Structural insights into the role of mutations in amyloidogenesis. J Biol Chem. 2008;283(44):30950-30956. doi:10.1074/jbc.M804822200
-
Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016;23(4):209-213. doi:10.1080/13506129.2016.1257986
-
Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22(18):3751-3757. doi:10.1200/JCO.2004.03.029
-
Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134(4):545-551. doi:10.5858/134.4.545
-
Shi J, Guan J, Jiang B, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38α MAPK pathway. Proc Natl Acad Sci USA. 2010;107(9):4188-4193. doi:10.1073/pnas.0912263107
-
Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis: recognition, confirmation, prognosis, and therapy. Mayo Clin Proc. 1999;74(5):490-494. doi:10.4065/74.5.490
-
Conceição I, González-Duarte A, Obici L, et al. "Red-flag" symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5-9. doi:10.1111/jns.12153
-
Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-2506. doi:10.1182/blood-2002-07-2299
-
Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am J Hematol. 2005;79(4):319-328. doi:10.1002/ajh.20381
-
Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015;168(2):207-218. doi:10.1111/bjh.13156
-
Vrana JA, Gamez JD, Madden BJ, et al. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114(24):4957-4959. doi:10.1182/blood-2009-07-230722
-
Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22(18):3751-3757. doi:10.1200/JCO.2004.03.029
-
Bochtler T, Hegenbart U, Kunz C, et al. Translocation t(11;14) is associated with adverse outcome in patients with newly diagnosed AL amyloidosis when treated with bortezomib-based regimens. J Clin Oncol. 2015;33(12):1371-1378. doi:10.1200/JCO.2014.57.4947
-
Hawkins PN, Lavender JP, Pepys MB. Evaluation of systemic amyloidosis by scintigraphy with 123I-labeled serum amyloid P component. N Engl J Med. 1990;323(8):508-513. doi:10.1056/NEJM199008233230803
-
Kastritis E, Palladini G, Minnema MC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385(1):46-58. doi:10.1056/NEJMoa2028631
-
Palladini G, Sachchithanantham S, Milani P, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126(5):612-615. doi:10.1182/blood-2015-01-620302
-
Reece DE, Hegenbart U, Sanchorawala V, et al. Efficacy and safety of once-weekly and twice-weekly bortezomib in patients with relapsed systemic AL amyloidosis: results of a phase 1/2 study. Blood. 2011;118(4):865-873. doi:10.1182/blood-2011-02-334227
-
D'Souza A, Dispenzieri A, Wirk B, et al. Improved outcomes after autologous hematopoietic cell transplantation for light chain amyloidosis: a Center for International Blood and Marrow Transplant Research study. J Clin Oncol. 2015;33(32):3741-3749. doi:10.1200/JCO.2015.62.4015
-
Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541-4549. doi:10.1200/JCO.2011.37.7614
-
Sidiqi MH, Aljama MA, Buadi FK, et al. Venetoclax for the treatment of translocation (11;14) AL amyloidosis. Blood Cancer J. 2020;10(5):55. doi:10.1038/s41408-020-0321-6
-
Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation. 1981;63(6):1285-1288. doi:10.1161/01.cir.63.6.1285
-
Herrmann SM, Gertz MA, Stegall MD, et al. Long-term outcomes of patients with light chain amyloidosis (AL) after renal transplantation with or without stem cell transplantation. Nephrol Dial Transplant. 2011;26(6):2032-2036. doi:10.1093/ndt/gfq683
-
Edwards CV, Rao N, Bhutani D, et al. Phase 1a/b study of monoclonal antibody CAEL-101 (11-1F4) in patients with AL amyloidosis. Blood. 2021;138(25):2632-2641. doi:10.1182/blood.2021011737
Last Updated: 2026-01-05
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Multiple Myeloma
- Monoclonal Gammopathy of Undetermined Significance (MGUS)
Differentials
Competing diagnoses and look-alikes to compare.
- AA Amyloidosis
- ATTR Amyloidosis (Hereditary and Wild-Type)
- Dialysis-Related Amyloidosis (Aβ2M)
Consequences
Complications and downstream problems to keep in mind.
- Heart Failure with Preserved Ejection Fraction
- Nephrotic Syndrome
- Restrictive Cardiomyopathy