Angelman Syndrome
Angelman syndrome (AS) is a rare neurogenetic disorder caused by loss of function of the maternally inherited UBE3A gene... MRCPCH exam preparation.
What matters first
Angelman syndrome (AS) is a rare neurogenetic disorder caused by loss of function of the maternally inherited UBE3A gene... MRCPCH exam preparation.
Seizures (common and often refractory) — may require polytherapy
9 Jan 2025
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Seizures (common and often refractory) — may require polytherapy
- Status epilepticus — emergency management
- Severe sleep disturbance impacting family
- Swallowing difficulties/aspiration risk
Exam focus
Current exam surfaces linked to this topic.
- MRCPCH
Linked comparisons
Differentials and adjacent topics worth opening next.
- Prader-Willi Syndrome
- Rett Syndrome
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Angelman Syndrome
1. Clinical Overview
Summary
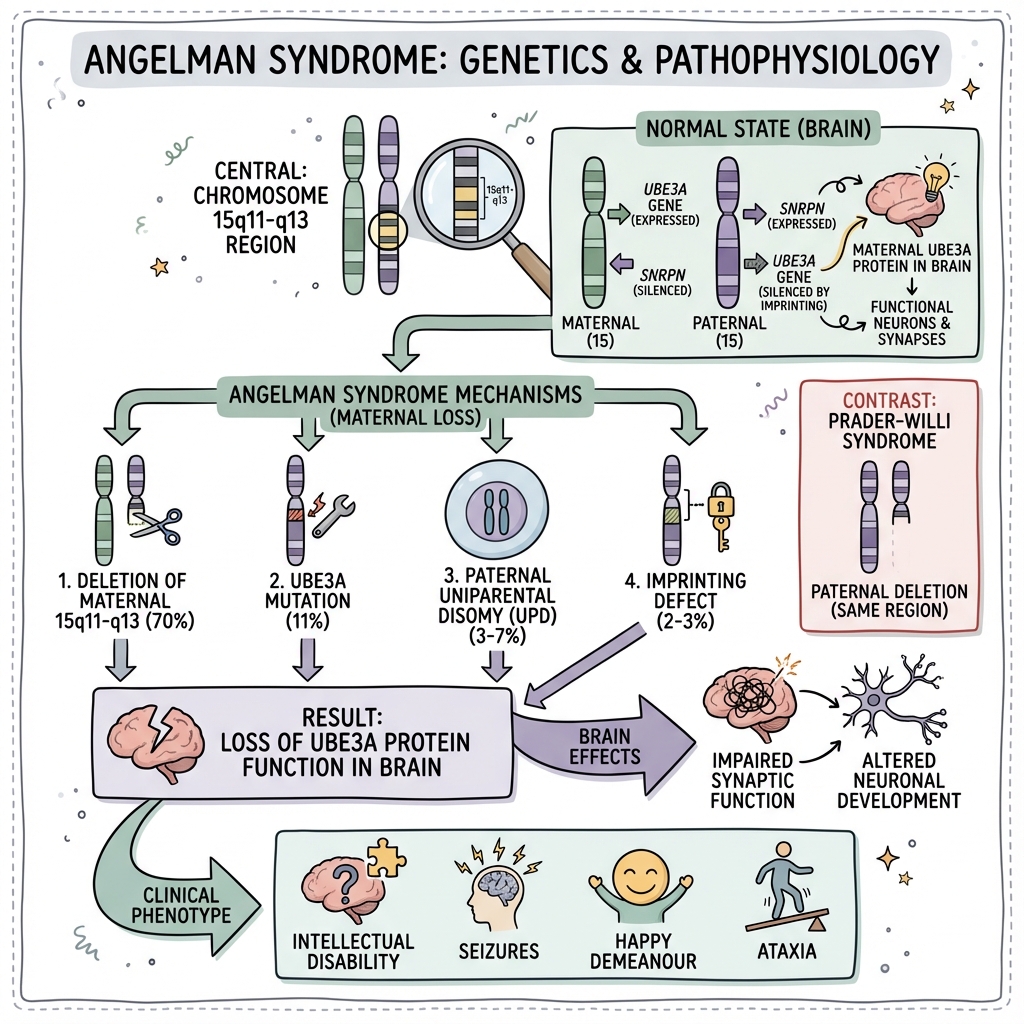
Angelman syndrome (AS) is a rare neurogenetic disorder caused by loss of function of the maternally inherited UBE3A gene on chromosome 15q11.2-q13.1. First described by British paediatrician Harry Angelman in 1965, the syndrome is characterised by a distinctive constellation of features including severe intellectual disability, absent or severely limited speech, movement disorders (ataxia, tremor, jerky limb movements), characteristic behaviours (frequent laughter, happy demeanour, hand-flapping, fascination with water), and seizures that are often treatment-resistant. [1,2]
The condition results from a unique phenomenon called genomic imprinting, whereby certain genes are expressed exclusively from one parental allele. In neurons, UBE3A is expressed almost exclusively from the maternal chromosome, while the paternal allele is epigenetically silenced. Consequently, loss of the maternal UBE3A copy leaves neurons without functional UBE3A protein, resulting in the characteristic neurological phenotype. This same chromosomal region (15q11.2-q13.1), when affected on the paternal chromosome, causes the clinically distinct Prader-Willi syndrome. [3,4]
The estimated prevalence is 1 in 12,000 to 1 in 20,000 live births, with equal distribution between sexes and across all ethnic groups. Diagnosis is confirmed through genetic testing, typically using DNA methylation analysis as the first-line test. Management is supportive and multidisciplinary, focusing on seizure control, communication strategies (augmentative and alternative communication), developmental intervention, sleep management, and comprehensive family support. While affected individuals require lifelong care, those with appropriate support can live into adulthood with near-normal life expectancy and meaningful quality of life. [5,6]
Key Facts
| Domain | Details |
|---|---|
| Definition | Neurogenetic disorder from loss of maternal UBE3A gene function at chromosome 15q11.2-q13.1 |
| Inheritance | Imprinting disorder; majority sporadic (70% deletion); variable recurrence risk by mechanism |
| Prevalence | 1:12,000 to 1:20,000 live births |
| Sex Distribution | Equal (male:female 1:1) |
| Age at Diagnosis | Typically 3-7 years; may be earlier with increased awareness |
| Life Expectancy | Near-normal with appropriate care |
| Key Gene | UBE3A (ubiquitin protein ligase E3A) |
| Chromosome Location | 15q11.2-q13.1 (imprinted region) |
Key Mnemonic
"A for Angelman, A for mAternAl": Angelman syndrome results from loss of the MATERNAL copy of the UBE3A gene. Contrast with Prader-Willi syndrome (Paternal loss) — same region, opposite parent.
Clinical Pearls
"Happy Puppet" Terminology — Deprecated: The historic term "Happy Puppet Syndrome" is considered offensive and stigmatising. Use "Angelman syndrome" exclusively.
Seizure Medication Warning: Carbamazepine, oxcarbazepine, vigabatrin, and tiagabine can worsen seizures in Angelman syndrome. Valproate, levetiracetam, and clobazam are typically first-line agents. [7]
EEG Pattern — Diagnostic Clue: Characteristic high-amplitude slow (2-3 Hz) delta activity with intermittent rhythmic theta over posterior regions is highly suggestive of AS, even before clinical seizures manifest. [8]
Deletion Size Matters: Larger deletions (Class I: BP1-BP3) may have more severe phenotypes than smaller deletions (Class II: BP2-BP3), though significant phenotypic variability exists. [9]
UPD and Imprinting Defects — Milder Phenotype: Patients with paternal uniparental disomy (UPD) or imprinting centre defects often have milder presentations with better expressive language and fewer seizures compared to deletion patients. [10]
Why This Matters Clinically
Angelman syndrome requires early recognition for appropriate developmental support, seizure management, and genetic counselling. The distinctive behavioural phenotype (happy demeanour, frequent laughter) combined with developmental delay can sometimes paradoxically delay diagnosis in early infancy when these "positive" behaviours mask underlying neurological impairment. Accurate genetic diagnosis is essential for determining recurrence risk, which varies dramatically by molecular mechanism (from less than 1% for de novo deletion to 50% for inherited UBE3A mutations). With appropriate support, individuals with AS can lead fulfilling lives, though the need for lifelong care places significant demands on families requiring comprehensive support services.
2. Epidemiology
Incidence & Prevalence
| Measure | Value | Evidence Source |
|---|---|---|
| Prevalence | 1 in 12,000 to 1 in 20,000 live births | [5,6] |
| Annual Incidence | ~1:20,000 | [6] |
| Cumulative Incidence | Similar across populations | [5] |
| Trend | Increasing recognition with improved genetic testing | [11] |
Demographics
| Factor | Details |
|---|---|
| Sex | Equal distribution (1:1 male:female) |
| Ethnicity | No ethnic predilection; all populations affected |
| Geography | Worldwide distribution |
| Socioeconomic | No association with socioeconomic status |
| Parental Age | Maternal age not a significant risk factor (unlike some aneuploidies) |
Age Distribution
| Age Group | Clinical Features |
|---|---|
| 0-6 months | Often appear normal; subtle hypotonia, feeding difficulties may be present |
| 6-12 months | Developmental delay becomes more apparent; lack of babbling |
| 1-3 years | Classic features emerge; seizures typically begin; characteristic behaviours |
| 3-7 years | Peak age at diagnosis; full phenotype established |
| Adolescence | Seizures may improve; scoliosis develops; weight gain tendency |
| Adulthood | Stable phenotype; continued need for care; aging-related concerns emerging |
3. Aetiology & Pathophysiology
Genetic Mechanisms
Angelman syndrome results from loss of function of the maternally expressed UBE3A gene at chromosome 15q11.2-q13.1. The UBE3A gene encodes E6-AP ubiquitin ligase (also known as UBE3A or E6AP), an E3 ubiquitin-protein ligase crucial for protein degradation through the ubiquitin-proteasome pathway. [1,3]
Molecular Mechanisms of Disease
| Mechanism | Frequency | Description | Recurrence Risk |
|---|---|---|---|
| Maternal Deletion (de novo) | 70-75% | Large deletion (~5-7 Mb) of maternal 15q11.2-q13.1 including UBE3A and multiple flanking genes | less than 1% |
| Maternal Deletion (inherited) | Rare | Deletion inherited from mother who carries a balanced translocation | Up to 50% |
| UBE3A Point Mutation | 10-15% | Intragenic mutations (missense, nonsense, frameshift) in UBE3A gene | Up to 50% if mother is carrier |
| Paternal Uniparental Disomy (UPD) | 3-7% | Child inherits both chromosome 15s from father (no maternal contribution) | less than 1% |
| Imprinting Centre Defect (de novo) | 2-3% | Epimutation affecting the Angelman syndrome imprinting centre (AS-IC) | less than 1% |
| Imprinting Centre Defect (inherited) | less than 1% | Microdeletion or mutation of AS-IC | Up to 50% |
| Unknown Mechanism | 10-15% | Clinical diagnosis but negative molecular testing | Empiric 10% |
Deletion Classes
Large deletions are classified by breakpoint (BP) location:
| Class | Breakpoints | Size | Associated Features |
|---|---|---|---|
| Class I | BP1 to BP3 | ~5.9-6.6 Mb | Larger; may include NIPA1, NIPA2; possibly more severe phenotype [9] |
| Class II | BP2 to BP3 | ~5.3-5.8 Mb | More common; slightly milder than Class I |
| Atypical | Variable | Variable | Smaller or larger than typical; phenotype depends on genes involved |
Genomic Imprinting at 15q11.2-q13.1
The 15q11.2-q13.1 region contains multiple imprinted genes expressed from only one parental allele:
Paternally Expressed (Maternally Silenced):
- SNRPN (small nuclear ribonucleoprotein polypeptide N)
- SNURF
- Multiple snoRNA genes (HBII-52 cluster, HBII-85 cluster)
- NDN (necdin)
- MAGEL2
- MKRN3
- These genes are lost in Prader-Willi syndrome
Maternally Expressed (Paternally Silenced in Neurons):
- UBE3A — expressed from maternal allele in neurons only
- ATP10A (tissue-specific)
The tissue-specific imprinting of UBE3A is particularly important: UBE3A is biallelically expressed in most tissues, but in neurons, the paternal allele is silenced by a long non-coding antisense transcript (UBE3A-ATS) originating from the SNURF-SNRPN locus. This neuron-specific paternal silencing explains why loss of the maternal UBE3A allele causes neurological disease while other tissues remain largely unaffected. [3,4,12]
Pathophysiology
Step 1: Loss of Maternal UBE3A Function
Through any of the mechanisms described above, the maternally-derived functional UBE3A allele is lost. Since the paternal allele is silenced in neurons by UBE3A-ATS, neurons are left without functional UBE3A protein.
Step 2: Impaired Ubiquitin-Proteasome Function
UBE3A (E6-AP) is an E3 ubiquitin ligase that:
- Transfers ubiquitin to target proteins
- Marks proteins for degradation by the 26S proteasome
- Regulates levels of multiple neuronal proteins
Key UBE3A substrates include:
- Arc (activity-regulated cytoskeleton-associated protein) — regulates AMPA receptor trafficking and synaptic plasticity [13]
- Sacsin — involved in mitochondrial dynamics
- Annexin A1 — role in inflammation and neuronal signalling
- p53 — tumour suppressor (in presence of HPV E6 protein)
Step 3: Synaptic Dysfunction
Loss of UBE3A leads to:
- Impaired synaptic plasticity — abnormal long-term potentiation (LTP)
- Altered dendritic spine morphology — immature spine phenotype
- Disrupted excitatory/inhibitory balance — cortical hyperexcitability
- Abnormal neuronal development — altered migration and differentiation
- Mitochondrial dysfunction — impaired energy metabolism [14]
Step 4: Clinical Phenotype
The neuronal dysfunction manifests as:
- Severe intellectual disability — learning and memory deficits
- Absent/minimal speech — expressive language severely affected
- Movement disorder — ataxia, tremor from cerebellar/motor pathway dysfunction
- Seizures — cortical hyperexcitability
- Sleep disturbance — disrupted circadian rhythm, reduced melatonin
- Characteristic behaviours — limbic system involvement
Genotype-Phenotype Correlations
| Mechanism | Phenotype Characteristics |
|---|---|
| Deletion | Most severe phenotype; hypopigmentation (OCA2 deletion); more severe seizures; microcephaly more common [9,10] |
| UBE3A Mutation | Variable severity; no hypopigmentation; may have better seizure control |
| Paternal UPD | Often milder; better expressive language (may develop single words); less severe seizures; higher BMI [10] |
| Imprinting Defect | Similar to UPD; relatively milder phenotype; better language outcomes |
4. Clinical Presentation
Natural History by Age
Prenatal Period
- Usually normal prenatal course
- No consistent ultrasound findings
- Normal birth weight and APGAR scores typical
Infancy (0-12 months)
| Feature | Timing | Frequency |
|---|---|---|
| Feeding difficulties (weak suck, GORD) | Birth onwards | 70-80% |
| Hypotonia (truncal) | Birth onwards | 60-70% |
| Developmental delay (subtle) | 4-6 months | Often not recognised |
| Sleep disturbance | 4-6 months onwards | 70-80% |
| Social smiling (often preserved) | 2-3 months | Normal timing |
| Tongue thrusting | 6+ months | Common |
| Drooling | 6+ months | Very common |
Early Childhood (1-4 years)
| Feature | Timing | Frequency |
|---|---|---|
| Developmental delay (obvious) | By 12 months | 100% |
| Absent/minimal speech | By 2 years | 100% |
| Gait abnormality (ataxic, wide-based) | Walking age (24-48 months) | 100% |
| Seizures | Usually by age 3 | 80-90% |
| Characteristic behaviours emerge | 1-3 years | 100% |
| Microcephaly (acquired) | By 2 years | 80% |
| Hand-flapping/mouthing | 1-2 years | Very common |
| Happy demeanour/frequent laughter | 1-3 years | 100% |
| Fascination with water/mirrors | Variable | Very common |
Later Childhood (5-12 years)
| Feature | Notes |
|---|---|
| Seizures | May be frequent and difficult to control |
| Hyperactivity/short attention span | Peak in childhood |
| Sleep disturbance | Persists; reduced total sleep |
| Communication | Non-verbal communication may develop; AAC beneficial |
| Mobility | Most walk independently with ataxic gait |
| Puberty | Usually normal timing |
Adolescence and Adulthood
| Feature | Notes |
|---|---|
| Seizures | Often improve; some become seizure-free |
| Scoliosis | Develops in 40-70%; may require bracing/surgery |
| Weight gain | Common; tendency toward obesity |
| Behaviour | May become calmer; anxiety can emerge |
| Constipation | Chronic; requires ongoing management |
| Mobility | May decline; increased spasticity possible |
| Sleep | Continues to be problematic for many |
Cardinal Clinical Features
Consistent Features (100% of Patients)
-
Severe Developmental Delay
- IQ typically 20-40
- Functional level remains below 24-30 months
- Learning continues throughout life, albeit slowly
-
Movement Disorder
- Ataxic, wide-based, stiff-legged gait
- Jerky, puppet-like movements of limbs
- Tremulous movement of extremities (action tremor)
- Arms often held up and flexed, especially when walking or excited
- Hypermotoric, hyperkinetic behaviour
-
Speech Impairment
- Absent or minimal expressive language (typically less than 6 words)
- Receptive language better than expressive
- Non-verbal communication often well developed (gestures, eye pointing)
- May use augmentative communication devices successfully
-
Behavioural Phenotype
- Happy, excitable demeanour
- Frequent smiling and laughter (often unprovoked)
- Hand-flapping when excited
- Easily entertained; love of social interaction
- Short attention span
- Mouthing behaviours
- Attraction to/fascination with water, shiny objects, mirrors
Frequent Features (> 80% of Patients)
-
Microcephaly
- Typically acquired (normal at birth)
- Deceleration of head growth by age 2 years
- Absolute or relative microcephaly by age 2
-
Seizures
- Onset typically between 1-3 years (can be later)
- Multiple seizure types common (see Seizure Section)
- Often difficult to control
- May improve in adolescence/adulthood
-
Abnormal EEG
- Characteristic pattern even without clinical seizures
- High-amplitude slow spike-wave activity (2-3 Hz)
- Rhythmic 4-6 Hz theta activity (often frontal)
- Runs of high-amplitude 2-3 Hz activity posteriorly
- Triphasic waves [8]
Associated Features (20-80% of Patients)
| Feature | Frequency | Notes |
|---|---|---|
| Hypopigmentation | 70-80% (deletion only) | Relative to family; due to OCA2 gene deletion |
| Fair hair, skin, light eyes | 70-80% (deletion only) | OCA2 involvement |
| Wide mouth | 70-80% | Characteristic facial feature |
| Widely-spaced teeth | 60-70% | May have dental crowding later |
| Tongue thrusting | 80-90% | Oral-motor dysfunction |
| Feeding difficulties | 70-80% | Especially infancy |
| Drooling | 80-90% | Oral-motor dysfunction |
| Sleep disturbance | 70-80% | Reduced total sleep; frequent waking |
| Strabismus | 30-40% | Esotropia most common |
| Prognathism | 40-50% | More prominent with age |
| Scoliosis | 40-70% (adulthood) | Progressive; requires monitoring |
| Constipation | 70-80% | Chronic; often requires treatment |
| Truncal hypotonia | 60-70% | May evolve to increased limb tone |
| Hyperactive deep tendon reflexes | 50-60% | Upper motor neuron signs |
| Heat sensitivity | 50-60% | Impaired thermoregulation |
Facial Features
The facial gestalt in Angelman syndrome becomes more apparent with age:
| Age | Features |
|---|---|
| Infancy | Often non-dysmorphic; may appear normal |
| Childhood | Wide mouth; widely-spaced teeth; flat occiput; tongue thrusting |
| Adolescence/Adulthood | Prominent mandible (prognathism); pointed chin; deep-set eyes; wide mouth |
Additional craniofacial features:
- Flat occiput
- Midface hypoplasia (mild)
- Wide nasal base
- Upslanting palpebral fissures (variable)
- Brachycephaly
Seizure Characteristics
Seizures occur in 80-90% of individuals with AS and are a major cause of morbidity. [7,15]
Seizure Types
| Seizure Type | Frequency | Characteristics |
|---|---|---|
| Atypical Absence | Very common | Brief staring, eyelid flutter, may be subtle |
| Myoclonic | Very common | Brief jerks; may be subtle or severe |
| Generalised Tonic-Clonic | Common | Bilateral tonic-clonic activity |
| Atonic | Less common | Drop attacks; fall risk |
| Non-convulsive Status Epilepticus | Common | May be underrecognised; prolonged confusion/regression |
| Convulsive Status Epilepticus | Common | Medical emergency; AS patients at higher risk |
EEG Characteristics
The EEG in Angelman syndrome shows characteristic abnormalities that can support diagnosis even before clinical seizures: [8]
| Pattern | Description | Clinical Significance |
|---|---|---|
| High-amplitude 2-3 Hz delta | Runs of high-amplitude slow activity, often frontal predominance | Present in > 80%; often appears by age 2 |
| Rhythmic 4-6 Hz theta | Seen posteriorly; may be notched | Characteristic pattern |
| Triphasic waves | Sharp waves with triphasic morphology | Less specific but common |
| Spike-wave discharges | 2-3 Hz generalised spike-wave | Correlates with clinical seizures |
| Photoparoxysmal response | Abnormal response to photic stimulation | Common |
Red Flags
[!CAUTION] Red Flags — Urgent Attention Required:
- Status epilepticus — Common in AS; requires emergency management
- Prolonged seizures (> 5 minutes) — Low threshold for rescue medication
- Non-convulsive status — Consider if prolonged confusion/regression
- Aspiration/choking — Swallowing difficulties, especially liquids
- Severe self-injury — May occur during behavioural dysregulation
- Extreme sleep disturbance — Impacts child and family health
- Hyperthermia — Impaired thermoregulation; risk in hot weather
- Constipation with distension — Risk of impaction
- New onset regression — Consider other causes (seizures, metabolic)
5. Differential Diagnosis
Primary Differentials
| Condition | Key Distinguishing Features | Testing |
|---|---|---|
| Prader-Willi Syndrome | Paternal deletion/maternal UPD; hypotonia/poor feeding → hyperphagia/obesity; less severe ID; speech present (though delayed); no seizures typical; hypogonadism | Methylation testing (distinguishes PWS from AS) |
| Rett Syndrome | Females only; normal early development → regression 6-18 months; loss of hand skills; stereotyped hand movements; gait apraxia; MECP2 mutation | MECP2 gene testing |
| Pitt-Hopkins Syndrome | Severe ID; absent speech; episodic hyperventilation/breath-holding; distinctive facies (wide mouth, thick lips); TCF4 mutation | TCF4 gene testing |
| Mowat-Wilson Syndrome | Severe ID; absent speech; distinctive facies (uplifted earlobes, hypertelorism); Hirschsprung disease; ZEB2 mutation | ZEB2 gene testing |
| Christianson Syndrome (X-linked) | Males; similar to AS (ataxia, seizures, happy demeanour); progressive; SLC9A6 mutation | SLC9A6 gene testing |
| FOXG1 Syndrome | Severe ID; absent speech; microcephaly; seizures; stereotypies | FOXG1 gene testing |
| Kleefstra Syndrome | Moderate-severe ID; childhood hypotonia; distinctive facies; EHMT1 deletion/mutation | EHMT1 testing |
Comparison: Angelman Syndrome vs Prader-Willi Syndrome
| Feature | Angelman Syndrome | Prader-Willi Syndrome |
|---|---|---|
| Genetic Mechanism | Loss of MATERNAL 15q11-q13 (UBE3A) | Loss of PATERNAL 15q11-q13 (SNRPN, snoRNAs) |
| Birth/Infancy | Often appear normal; some hypotonia | Severe hypotonia; poor feeding |
| Intellectual Disability | Severe (IQ 20-40) | Mild-moderate (mean IQ 60-70) |
| Speech | Absent or less than 6 words | Delayed but usually develops |
| Motor | Ataxic gait; jerky movements | Gross motor delay; normal gait once walking |
| Behaviour | Happy, excitable, hand-flapping | Temper tantrums; food obsession; skin picking |
| Feeding | May have difficulties; no hyperphagia | Hyperphagia from age 2-8; obesity |
| Body Habitus | Usually normal or thin; obesity in adults | Characteristic obesity; small hands/feet |
| Seizures | Common (80%+) | Rare |
| Sleep | Reduced need for sleep; frequent waking | Excessive daytime sleepiness |
| Endocrine | Usually normal | Hypogonadism; growth hormone deficiency |
| Hypopigmentation | Yes (deletion cases) | Yes (deletion cases) |
When to Suspect Alternative Diagnosis
Consider alternative diagnosis if:
- Developmental regression (more typical of Rett, mitochondrial disorders)
- Early severe hypotonia with failure to thrive (PWS, myopathy)
- Normal EEG pattern in older child with suspected AS
- Male with X-linked inheritance pattern (Christianson syndrome)
- Hirschsprung disease present (Mowat-Wilson)
- Hyperventilation episodes (Pitt-Hopkins)
- Negative methylation testing and negative UBE3A sequencing
6. Clinical Examination
General Approach
A systematic examination focusing on key features of Angelman syndrome:
General Inspection
- Behaviour: Happy, smiling, excitable demeanour
- Interaction: Seeks social engagement; may approach examiner
- Movement: Hyperkinetic; hand-flapping; tremulous
- Posture: Arms may be held flexed and elevated
- Build: Usually normal; may trend toward obesity in adults
Growth Parameters
| Measurement | Expected Finding |
|---|---|
| Weight | Normal at birth; trend toward obesity in adulthood |
| Height | Usually normal |
| Head Circumference | Normal at birth; deceleration by age 2 years; microcephaly common |
Dysmorphic Examination
| Feature | Findings |
|---|---|
| Head Shape | Flat occiput; brachycephaly |
| Face | Midface hypoplasia (mild); deep-set eyes |
| Mouth | Wide; thin upper lip; widely-spaced teeth |
| Mandible | Prognathism (more prominent with age) |
| Tongue | Protrusion; thrusting |
| Hair/Skin/Eyes | Hypopigmentation relative to family (deletion cases) |
| Hands/Feet | Usually normal; may be small |
Neurological Examination
| Component | Findings |
|---|---|
| Tone (Trunk) | Hypotonia |
| Tone (Limbs) | May be normal, hypotonic, or increased with age |
| Power | Usually normal |
| Reflexes | Often brisk/hyperactive |
| Plantar Response | May be extensor |
| Gait | Wide-based, ataxic, stiff-legged; arms often elevated |
| Coordination | Ataxic; action tremor; intention tremor |
| Involuntary Movements | Jerky movements; tremor; myoclonus |
Communication Assessment
| Domain | Findings |
|---|---|
| Expressive Language | Absent or less than 6 words |
| Receptive Language | Better than expressive; follows simple commands |
| Non-verbal Communication | Often well-developed (gestures, pointing, leading) |
| Social Engagement | Typically good; eye contact maintained |
Specific Tests
| Test | Purpose | Findings |
|---|---|---|
| Head Circumference | Monitor for microcephaly | Often less than 3rd centile by age 2 |
| Gait Assessment | Characterise motor phenotype | Wide-based, ataxic, puppet-like |
| Hand Function | Fine motor assessment | Tremor; difficulty with fine tasks |
| Oral-Motor Examination | Assess feeding/swallowing | Tongue thrusting; drooling |
| Spine Examination | Scoliosis screening | Spinal curvature (especially adolescence) |
| Eye Examination | Assess for strabismus | Esotropia common |
7. Investigations
Diagnostic Testing Algorithm
Step 1: Clinical Suspicion ↓ Step 2: DNA Methylation Analysis (First-line)
- Abnormal → AS confirmed; proceed to characterise mechanism
- Normal → If strong suspicion, proceed to Step 5 ↓ Step 3: Determine Molecular Mechanism
- Chromosomal Microarray (CMA) or FISH → Detects deletion
- If no deletion → UPD testing (SNP array, microsatellite analysis)
- If no deletion or UPD → Imprinting centre analysis ↓ Step 4: Family Testing
- If deletion → Parental karyotype (rule out balanced translocation)
- If UBE3A mutation → Maternal testing for carrier status
- If imprinting defect → Characterise (deletion vs epimutation) ↓ Step 5: If Methylation Normal but Clinical Suspicion High
- UBE3A gene sequencing
- Consider alternate diagnoses if negative
Specific Investigations
Genetic Testing
| Test | Methodology | Detects | Sensitivity |
|---|---|---|---|
| DNA Methylation Analysis | Methylation-specific PCR (MS-PCR) or methylation-specific MLPA (MS-MLPA) | Abnormal methylation pattern in AS (all mechanisms except ~10% unknown) | ~80-85% overall; 100% for deletion, UPD, imprinting defects |
| Chromosomal Microarray (CMA) | Array CGH or SNP array | Deletions; also detects UPD (SNP array) | 100% for deletions |
| FISH | Fluorescence in situ hybridisation | 15q11.2-q13.1 deletion | 100% for deletions |
| UPD Analysis | SNP array or microsatellite markers | Paternal uniparental disomy | ~95-99% |
| UBE3A Sequencing | Sanger or NGS | Point mutations, small insertions/deletions | ~100% for UBE3A mutations |
| Imprinting Centre Analysis | MLPA, sequencing | IC deletions or mutations | ~95% |
Neurological Investigations
| Test | Indication | Expected Findings |
|---|---|---|
| EEG | All patients; seizure evaluation | Characteristic high-amplitude slow activity; spike-wave; may be abnormal before clinical seizures [8] |
| MRI Brain | Consider in all; required if atypical features | Usually normal or non-specific (mild atrophy, delayed myelination); structural abnormality rare |
| Video-EEG Monitoring | Characterise seizures; assess for non-convulsive status | Capture seizure types; quantify episodes |
Other Investigations
| Test | Purpose | When to Perform |
|---|---|---|
| Sleep Study (Polysomnography) | Characterise sleep architecture; rule out sleep apnoea | Significant sleep disturbance; snoring; obesity |
| Swallowing Assessment (VFSS/FEES) | Evaluate swallowing safety | Recurrent chest infections; coughing with feeds; drooling |
| Spinal X-ray | Scoliosis screening | Adolescence; clinical suspicion of curvature |
| Ophthalmology Assessment | Strabismus; refractive error | All patients |
| Audiology | Hearing assessment | All patients |
| DEXA Scan | Bone density | Consider if immobile or on long-term antiepileptics |
| Dental Assessment | Oral health; malocclusion | Regular assessment |
8. Classification
Molecular Classification
| Class | Mechanism | Frequency | Recurrence Risk | Phenotype Notes |
|---|---|---|---|---|
| Class I | De novo deletion | 70% | less than 1% | Most common; typical severe phenotype |
| Class II | UBE3A mutation | 11% | Up to 50% (if maternal carrier) | Variable severity |
| Class III | Paternal UPD | 3-7% | less than 1% | Often milder phenotype [10] |
| Class IV | Imprinting centre defect | 2-3% | Variable (up to 50% if inherited IC deletion) | Often milder phenotype |
| Class V | Unknown mechanism | 10-15% | Empiric ~10% | Clinical diagnosis; molecular etiology unclear |
Deletion Size Classification
| Type | Breakpoints | Approximate Size | Genes Involved | Clinical Notes |
|---|---|---|---|---|
| Class I Deletion | BP1-BP3 | 5.9-6.6 Mb | NIPA1, NIPA2, CYFIP1, TUBGCP5 + critical region | Larger; may be more severe [9] |
| Class II Deletion | BP2-BP3 | 5.3-5.8 Mb | Critical region only | Most common deletion class |
| Atypical | Variable | Variable | Variable | Phenotype depends on genes involved |
Severity Classification (Functional)
No validated severity classification exists, but functional domains can be assessed:
| Domain | Mild | Moderate | Severe |
|---|---|---|---|
| Mobility | Independent ambulation, minimal ataxia | Independent ambulation with ataxic gait | Wheelchair dependent or non-ambulatory |
| Communication | Uses 5+ words or proficient AAC | Uses 1-5 words or basic AAC | No words; minimal AAC use |
| Seizure Control | Seizure-free or rare seizures | Seizures controlled on medication | Refractory seizures |
| Self-Care | Some independence in daily activities | Requires moderate assistance | Fully dependent for all care |
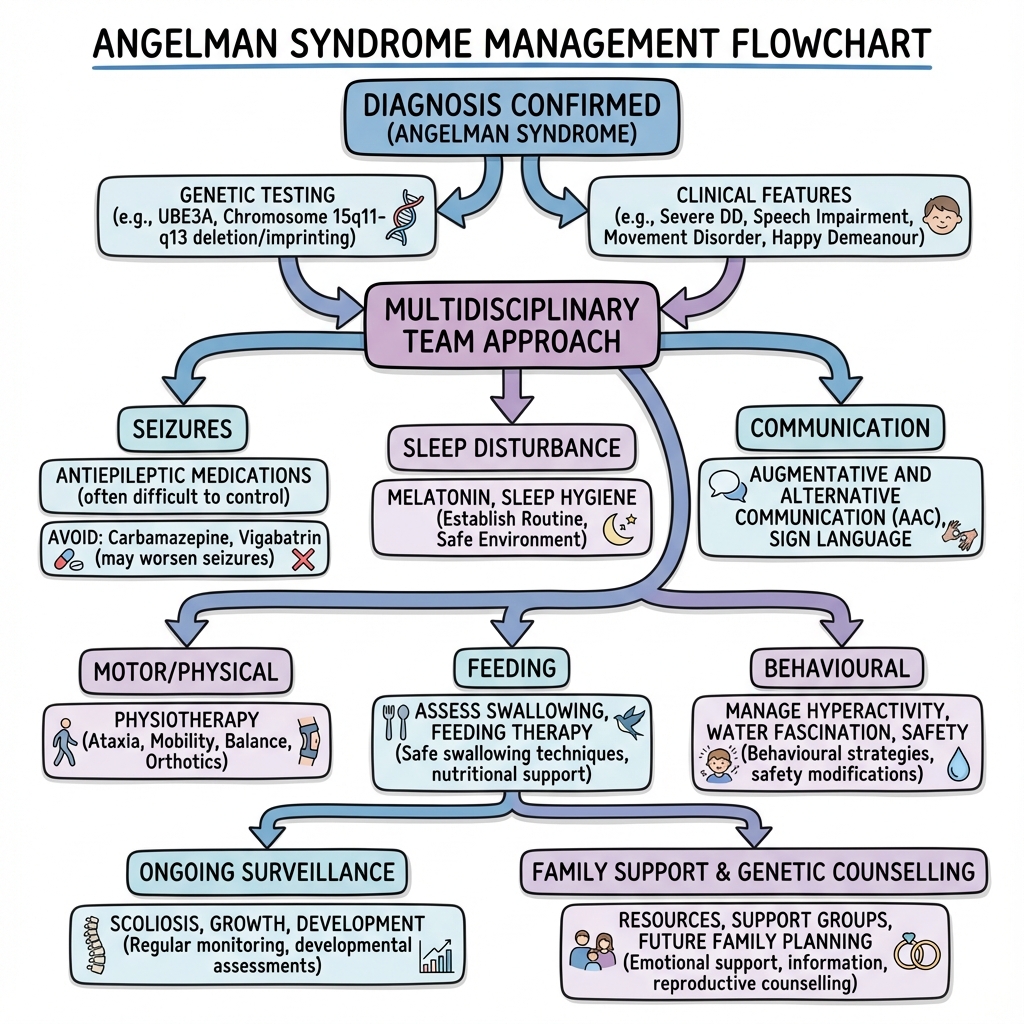
9. Management
Management Principles
- Multidisciplinary team approach — essential for comprehensive care
- Early intervention — optimise developmental potential
- Seizure control — major impact on quality of life
- Communication support — AAC enables meaningful participation
- Family-centred care — support for whole family
- Transition planning — prepare for adult services
Multidisciplinary Team
| Team Member | Role |
|---|---|
| Paediatric Neurologist | Seizure management; EEG interpretation; medication optimisation |
| Clinical Geneticist | Diagnosis confirmation; family counselling; recurrence risk |
| Developmental Paediatrician | Developmental assessment; care coordination; behavioural support |
| Speech and Language Therapist | AAC implementation; feeding/swallowing; communication strategies |
| Physiotherapist | Motor development; mobility; scoliosis prevention; equipment |
| Occupational Therapist | Daily living skills; sensory processing; environmental adaptation |
| Dietitian | Nutrition; constipation management; obesity prevention |
| Sleep Medicine | Sleep disturbance evaluation and management |
| Psychologist | Behavioural support; family support; mental health |
| Social Worker | Family support; benefits; respite; transition planning |
| Special Education Specialist | Educational planning; school support |
| Orthopaedic Surgeon | Scoliosis management; hip surveillance |
| Ophthalmologist | Strabismus; refractive error |
| Dentist | Oral health; malocclusion; drooling management |
Seizure Management
Pharmacological Management
| Agent | Role | Notes |
|---|---|---|
| Valproate (Sodium Valproate) | First-line; broad-spectrum | Effective for multiple seizure types; monitor LFTs; teratogenic (counsel) [7,15] |
| Levetiracetam | First-line alternative | Broad-spectrum; may worsen behaviour in some patients |
| Clobazam | First-line or adjunctive | Useful for multiple seizure types; tolerance may develop |
| Clonazepam | Adjunctive | Myoclonic seizures; tolerance develops |
| Lamotrigine | Second-line | May be useful; high doses can worsen myoclonic seizures |
| Topiramate | Second-line | May help; weight loss side effect can be problematic |
| Ethosuximide | Absence seizures | Specific for typical absence component |
| Phenobarbital | Refractory cases | Sedation; cognitive effects |
Medications to AVOID
[!WARNING] Contraindicated or Use with Extreme Caution:
- Carbamazepine — Can worsen myoclonic and absence seizures
- Oxcarbazepine — Same concerns as carbamazepine
- Vigabatrin — Can worsen seizures in AS
- Tiagabine — May worsen seizures
- Phenytoin — Generally avoid in generalised epilepsy syndromes
- Gabapentin/Pregabalin — May worsen myoclonic seizures
Non-Pharmacological Approaches
| Approach | Evidence | Notes |
|---|---|---|
| Ketogenic Diet | Moderate evidence | Consider for refractory seizures; requires dietitian support; may improve alertness [16] |
| Modified Atkins Diet | Limited evidence | Easier to implement than classic ketogenic diet |
| Vagus Nerve Stimulation (VNS) | Case series support | Consider for refractory cases; may reduce seizure frequency |
| Corpus Callosotomy | Rare indication | Only for severe drop attacks refractory to all else |
Status Epilepticus Management
- Follow standard paediatric status epilepticus protocols
- AS patients may have prolonged seizures — low threshold for rescue medication
- Parents should have emergency rescue medication (buccal midazolam, rectal diazepam)
- Hospital protocol should be documented for emergency presentations
Sleep Management
| Intervention | Details | Evidence |
|---|---|---|
| Sleep Hygiene | Regular bedtime; dark, cool room; reduce stimulation before bed; consistent routine | Expert consensus |
| Melatonin | 2.5-10 mg at bedtime; first-line pharmacological | Good evidence; commonly effective [17] |
| Behavioural Strategies | Extinction techniques; positive bedtime routines | Variable success |
| Address Underlying Causes | Pain, constipation, GORD, seizures | Important to exclude |
| Consider Sleep Study | If sleep apnoea suspected (obesity, snoring, witnessed apnoeas) | Standard approach |
| Iron Supplementation | If ferritin less than 50 μg/L (restless legs) | May help sleep quality |
Communication and Development
Augmentative and Alternative Communication (AAC)
| Type | Examples | Notes |
|---|---|---|
| Low-Tech | Picture Exchange Communication System (PECS); communication boards; symbol charts | Start early; build vocabulary progressively |
| High-Tech | Speech-generating devices (SGD); tablet-based apps | Many individuals can use touchscreens effectively |
| Sign Language | Makaton; Baby Sign; BSL | Many can learn simple signs; complements other AAC |
| Eye Gaze | Eye gaze technology | For those with limited hand function |
Key Principles:
- Start AAC early — do not wait for speech to "fail"
- Multimodal approach — combine different AAC methods
- Total communication — accept all forms of communication
- Consistent use across settings (home, school, community)
- Regular review and vocabulary expansion
Early Intervention
| Domain | Approach |
|---|---|
| Motor Development | Physiotherapy; occupational therapy; hydrotherapy |
| Cognitive Development | Structured teaching; visual supports; repetition |
| Social Skills | Social stories; group activities; peer interaction |
| Adaptive Behaviour | Daily living skills training; consistent routines |
Behavioural Management
| Behaviour | Approach |
|---|---|
| Hyperactivity | Structured environment; regular routine; physical activity outlets |
| Short Attention Span | Brief, engaging activities; visual supports; frequent breaks |
| Mouthing Behaviours | Provide safe alternatives; redirect; sensory input |
| Sleep-Related Behaviours | Consistent routine; melatonin; safe bedroom environment |
| Anxiety (emerging with age) | Predictable routines; social stories; consider behavioural approaches |
| Aggression (rare) | Identify triggers; functional behaviour analysis; positive behaviour support |
Safety Management
| Risk | Management |
|---|---|
| Water Fascination | Supervise near all water sources; pool fencing; water alarms |
| Lack of Danger Awareness | Secure environment; locks on doors/windows; GPS tracking |
| Wandering/Elopement | Secure doors; alarms; ID bracelet; community awareness |
| Heat Sensitivity | Avoid overheating; air conditioning; monitor in hot weather |
| Seizures | Rescue medication available; seizure action plan; alert bracelet |
Feeding and Nutrition
| Issue | Management |
|---|---|
| Feeding Difficulties (Infancy) | Occupational therapy; positioning; texture modification |
| Drooling | Oral-motor therapy; anticholinergics (glycopyrrolate); botulinum toxin; surgery (rare) |
| Constipation | Adequate fluid; dietary fibre; regular toileting; laxatives (movicol, lactulose) |
| Obesity (Adult) | Dietitian involvement; portion control; physical activity; monitor regularly |
| GORD | Positioning; thickened feeds; PPIs if indicated |
Orthopaedic Management
| Issue | Management |
|---|---|
| Scoliosis | Clinical monitoring from adolescence; spinal X-ray if concerns; bracing; surgery if > 40-50° or progressive |
| Hip Dysplasia | Clinical and radiological surveillance in non-ambulatory individuals |
| Contractures | Physiotherapy; stretching; splinting; orthotics |
| Osteoporosis | Weight-bearing activity; vitamin D; calcium; DEXA if concern |
Ongoing Surveillance
| Issue | Frequency | Notes |
|---|---|---|
| Neurology Review | 6-12 monthly | Seizure control; medication optimisation |
| Developmental Assessment | Annually | Track progress; adjust interventions |
| Scoliosis Screening | Annually from age 10; earlier if concerns | Clinical examination; X-ray if curvature |
| Weight Monitoring | Each visit | Trend toward obesity; intervene early |
| Constipation Review | Each visit | Common; prevent complications |
| Sleep Assessment | Each visit | Major quality of life issue |
| Vision/Hearing | Annually | Strabismus; refractive errors; hearing |
| Dental Review | 6-12 monthly | Oral health; drooling; malocclusion |
| Transition Planning | Start by age 14 | Prepare for adult services |
Transition to Adult Services
| Age | Actions |
|---|---|
| 14 years | Begin transition planning discussions; identify adult services |
| 16 years | Meet adult providers; begin capacity assessments; guardianship considerations |
| 18 years | Transfer to adult services; legal/financial planning completed; day services in place |
Key transition considerations:
- Identify adult neurology/neurodevelopmental services
- Legal decision-making arrangements (guardianship/power of attorney)
- Financial planning (benefits, trusts)
- Housing and day services
- Ongoing medical management plan
- Emergency protocols
10. Complications
Medical Complications
| Complication | Frequency | Risk Factors | Management |
|---|---|---|---|
| Seizures | 80-90% | All genotypes; deletion may be more severe | Antiepileptic therapy; avoid contraindicated drugs |
| Status Epilepticus | Common | All patients with seizures | Emergency protocol; rescue medication |
| Scoliosis | 40-70% (adolescence/adulthood) | Hypotonia; immobility | Monitoring; bracing; surgery if severe |
| Constipation | 70-80% | Hypotonia; diet; medications | Dietary measures; laxatives |
| GORD | 40-60% | Hypotonia; feeding difficulties | PPIs; positioning; surgery (rarely) |
| Aspiration Pneumonia | Variable | Swallowing dysfunction; drooling | Swallowing assessment; thickened fluids; positioning |
| Obesity | Common (adults) | Reduced activity; diet | Dietitian; exercise; monitoring |
| Strabismus | 30-40% | Neurological | Ophthalmology; patching; surgery |
| Sleep Apnoea | Uncommon | Obesity; hypotonia | Sleep study; CPAP if confirmed |
| Hyperthermia | Variable | Impaired thermoregulation | Avoid overheating; air conditioning |
Developmental/Behavioural Complications
| Issue | Impact | Management |
|---|---|---|
| Severe Intellectual Disability | Lifelong learning needs; functional limitations | Ongoing education; adaptive support |
| Absent/Minimal Speech | Communication barrier | AAC; total communication approach |
| Sleep Disturbance | Impacts child and family | Sleep hygiene; melatonin; behavioural approaches |
| Hyperactivity/Short Attention | Impacts learning and safety | Structured environment; occupational therapy |
| Anxiety (emerging with age) | Quality of life impact | Routine; predictability; behavioural support |
Complications by Age
| Age | Key Complications |
|---|---|
| Infancy | Feeding difficulties; GORD; failure to thrive |
| Early Childhood | Seizure onset; developmental concerns; sleep problems |
| Later Childhood | Seizure control challenges; behavioural challenges |
| Adolescence | Scoliosis; seizure changes; puberty; transition |
| Adulthood | Obesity; reduced mobility; constipation; aging-related issues |
11. Prognosis & Outcomes
Natural History
Angelman syndrome is a lifelong condition with stable disability. There is no cognitive deterioration (unlike neurodegenerative conditions), and individuals continue to learn throughout life, although at a significantly slower pace. [18]
| Life Stage | Prognosis |
|---|---|
| Life Expectancy | Near-normal with appropriate care; some individuals live into 60s-70s |
| Seizures | May improve in adolescence/adulthood; some become seizure-free |
| Mobility | 75-90% achieve independent ambulation; may decline with age |
| Communication | Continues to develop through AAC; expressive speech rarely develops |
| Independence | Not anticipated; requires lifelong supervision and support |
| Quality of Life | Can have excellent quality of life with appropriate support |
Outcome by Domain
| Domain | Outcome | Notes |
|---|---|---|
| Cognitive | Severe ID persists; IQ typically 20-40 | Continues to learn throughout life |
| Motor | Most walk; gait remains ataxic; may decline with age | Falls remain a concern |
| Language | less than 6 words typical; AAC successful for many | Receptive > expressive |
| Social | Often excellent social engagement | Enjoys people; affectionate |
| Adaptive | Requires lifelong assistance | Self-care skills variable |
Prognostic Factors
Better Outcomes Associated With:
- Earlier diagnosis and intervention
- Effective seizure control
- Consistent AAC implementation
- Strong family and social support
- Access to specialised services
- Milder molecular mechanism (UPD, imprinting defect)
Poorer Outcomes Associated With:
- Refractory epilepsy
- Non-convulsive status epilepticus episodes
- Severe feeding and swallowing difficulties
- Recurrent aspiration pneumonia
- Severe scoliosis
- Limited access to services
- Larger deletions (Class I)
Emerging Therapies
Active areas of research with potential to modify the disease course: [19]
| Approach | Mechanism | Status |
|---|---|---|
| Antisense Oligonucleotides (ASOs) | Silence UBE3A-ATS to reactivate paternal UBE3A | Phase 1/2 clinical trials (GTX-102) |
| Gene Therapy | Deliver functional UBE3A gene | Preclinical |
| Small Molecule Activators | Unsilence paternal UBE3A | Preclinical |
| Artificial Transcription Factors | Activate paternal UBE3A | Preclinical |
[!NOTE] Research Hope: The theoretical possibility of "unsilencing" the intact paternal UBE3A allele offers a potential therapeutic approach that is unique among neurogenetic disorders. Multiple clinical trials are ongoing or planned.
12. Evidence & Guidelines
Key Clinical Guidelines
| Guideline | Source | Year | Key Recommendations |
|---|---|---|---|
| Angelman Syndrome Consensus Guidelines | Williams et al. | 2010 | Comprehensive clinical management; diagnostic criteria; surveillance recommendations [2] |
| Epilepsy Management in AS | Thibert et al. | 2009 | Antiepileptic drug recommendations; drugs to avoid [7] |
| Updated Diagnostic Criteria | Williams et al. | 2006 | Clinical diagnostic criteria; molecular testing algorithm [1] |
| Sleep Management | Pelc et al. | 2008 | Melatonin use; behavioural approaches [17] |
Landmark Studies
| Study | Year | Key Findings |
|---|---|---|
| Kishino et al.; Matsuura et al. | 1997 | Identification of UBE3A as the causative gene [3] |
| Buiting et al. | 1995 | Description of imprinting centre defects |
| Lossie et al. | 2001 | Genotype-phenotype correlations [10] |
| Margolis et al. | 2015 | UBE3A antisense transcript mechanism [12] |
| Meng et al. | 2013 | ASO approach to unsilence paternal UBE3A [19] |
Evidence Levels
| Intervention | Level of Evidence |
|---|---|
| Methylation testing for diagnosis | Level I |
| Valproate for seizures | Level III (consensus, cohort studies) |
| Avoid carbamazepine in AS | Level III (case series, consensus) |
| Melatonin for sleep | Level II (controlled studies) |
| AAC for communication | Level II (observational, outcomes research) |
| Ketogenic diet for refractory seizures | Level III (case series) |
13. Exam-Focused Content
Common Examination Questions
- "What are the genetic mechanisms underlying Angelman syndrome?"
- "How does Angelman syndrome differ from Prader-Willi syndrome?"
- "Describe the clinical features of Angelman syndrome."
- "What investigations would you order to confirm a diagnosis of Angelman syndrome?"
- "How would you manage seizures in a child with Angelman syndrome?"
- "What is the recurrence risk for Angelman syndrome?"
- "Explain genomic imprinting and its relevance to Angelman syndrome."
- "What are the characteristic EEG findings in Angelman syndrome?"
Viva Points
Viva Point: Opening Statement: "Angelman syndrome is a neurogenetic disorder caused by loss of function of the maternally-inherited UBE3A gene at chromosome 15q11.2-q13.1. It is characterised by severe intellectual disability, absent or minimal speech, ataxic movement disorder, seizures, and a characteristic happy demeanour with frequent laughter."
Key Facts to Cite:
- Prevalence 1:12,000 to 1:20,000 (Williams et al., 2010)
- 70% deletion, 11% UBE3A mutation, 3-7% paternal UPD, 2-3% imprinting defect
- Methylation testing first-line (80-85% sensitivity for all mechanisms)
- Seizures in 80-90%; avoid carbamazepine, vigabatrin
- Paternal silencing of UBE3A in neurons is key to pathophysiology
Model Answers
Q: A 3-year-old is referred with developmental delay, no speech, and an unusual happy demeanour with frequent laughter. What is your differential diagnosis and approach?
A: "This presentation is highly suggestive of Angelman syndrome, but I would consider a differential including other genetic causes of severe intellectual disability with absent speech, such as Rett syndrome, Pitt-Hopkins syndrome, Mowat-Wilson syndrome, and Christianson syndrome.
My approach would be:
- Detailed history: Pregnancy, birth, developmental trajectory, seizure history, sleep pattern, feeding
- Examination: Dysmorphic features, neurological examination focusing on tone, gait, movements
- Investigations: First-line DNA methylation testing (MS-PCR or MS-MLPA). If abnormal, confirm AS and determine mechanism with chromosomal microarray and if needed, UBE3A sequencing. EEG would show characteristic pattern even before clinical seizures.
If Angelman syndrome is confirmed, I would:
- Refer to genetics for family counselling
- Neurology for seizure management if present
- Start early intervention with speech therapy focusing on AAC
- Discuss with family regarding prognosis and support services"
Q: What antiepileptic drugs would you use and avoid in Angelman syndrome?
A: "Seizures in Angelman syndrome are often of multiple types including myoclonic, atypical absence, and generalised tonic-clonic, requiring broad-spectrum antiepileptic drugs.
First-line options:
- Sodium valproate — effective for multiple seizure types; monitor liver function
- Levetiracetam — broad-spectrum; may cause behavioural side effects in some
- Clobazam — useful adjunct; tolerance may develop
Avoid or use with caution:
- Carbamazepine and oxcarbazepine — can worsen myoclonic and absence seizures
- Vigabatrin — may worsen seizures
- Tiagabine — may worsen seizures
For refractory cases, consider ketogenic diet or vagus nerve stimulation. Parents should have rescue medication (buccal midazolam) as prolonged seizures are common."
Common Mistakes (Fail Points)
❌ Fails in examinations include:
- Confusing Angelman (maternal) with Prader-Willi (paternal) — must know imprinting correctly
- Recommending carbamazepine for seizures in AS
- Missing the characteristic EEG pattern as a diagnostic clue
- Not knowing recurrence risks vary by mechanism
- Failing to mention AAC for communication
- Not appreciating the range of molecular mechanisms beyond deletion
- Using the deprecated term "Happy Puppet Syndrome"
High-Yield Facts for Exams
| Topic | Key Point |
|---|---|
| Genetics | Maternal 15q11.2-q13.1 deletion (70%), UBE3A mutation (11%), paternal UPD (3-7%), imprinting defect (2-3%) |
| Imprinting | UBE3A paternally silenced in neurons only; other tissues biallelic |
| Comparison | Same region as PWS; AS = maternal loss, PWS = paternal loss |
| Presentation | Severe ID, no speech, ataxia, seizures, happy demeanour |
| Diagnosis | Methylation testing first-line; detects 80-85% |
| EEG | High-amplitude 2-3 Hz delta; rhythmic theta posteriorly |
| Seizure Drugs | Valproate, levetiracetam good; AVOID carbamazepine, vigabatrin |
| Prognosis | Lifelong care; near-normal life expectancy |
| Research | ASO therapy to unsilence paternal UBE3A in clinical trials |
14. Patient/Layperson Explanation
What is Angelman Syndrome?
Angelman syndrome is a rare genetic condition that affects the brain and nervous system. Children with Angelman syndrome have intellectual disability, trouble with movement and balance, difficulty speaking (most don't develop speech), and often have seizures (fits). One of the distinctive features is that affected children typically have a happy, excitable personality with frequent smiling and laughter.
Why Does It Happen?
Everyone has two copies of each gene — one from their mother and one from their father. Most genes work from both copies. However, for a small number of genes, only one copy is meant to be "switched on" depending on which parent it came from. This is called genomic imprinting.
The gene called UBE3A is one of these special genes. In brain cells, only the copy from the mother is switched on — the father's copy is naturally switched off. In Angelman syndrome, the mother's copy is missing or doesn't work. Because the father's copy is already switched off, brain cells have no working UBE3A, which causes the problems seen in the condition.
This is different from Prader-Willi syndrome, where the same area of the chromosome is affected, but it's the father's copy that's missing or not working — causing a completely different set of problems.
How is It Diagnosed?
Angelman syndrome is diagnosed with genetic tests, usually a special blood test that looks at the "imprinting pattern" of chromosome 15. This test can tell if the condition is present and often what type it is.
How is It Treated?
There is no cure for Angelman syndrome yet, but there are many ways to help:
-
Seizures: Medicines to control seizures. Some medicines that work for other types of epilepsy can actually make seizures worse in Angelman syndrome, so it's important to see a specialist.
-
Communication: Because speech is very limited, children are taught to communicate using sign language, picture symbols, or electronic devices with voices. This is called augmentative and alternative communication (AAC).
-
Physical therapy: To help with walking, balance, and movement.
-
Sleep: Many children with Angelman syndrome have trouble sleeping. Melatonin (a natural sleep hormone) and good sleep habits can help.
-
Support: Special education, therapy services, and support for the whole family are essential.
What to Expect
- Children with Angelman syndrome will need lifelong care and support
- With good care, most can live into adulthood
- Many learn to walk independently, though with an unusual gait
- Speech is very limited, but many communicate well using signs or devices
- Seizures often become easier to manage with age
- Despite challenges, people with Angelman syndrome often have a happy disposition and can enjoy life with their families
When to Seek Help
- If your child has a prolonged seizure (more than 5 minutes) — call 999/911
- If you notice new difficulties with swallowing or recurrent chest infections
- If sleep disturbance is severely affecting the family
- For genetic counselling if you are planning another pregnancy
Research and Hope
Scientists are working on new treatments that might help the brain switch on the father's copy of the UBE3A gene. This is an exciting area of research, and clinical trials are already happening. While there's no cure yet, there is real hope for treatments that could significantly improve outcomes in the future.
15. References
Primary Guidelines & Consensus
-
Williams CA, Beaudet AL, Clayton-Smith J, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140(5):413-418. doi:10.1002/ajmg.a.31074
-
Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385-395. doi:10.1097/GIM.0b013e3181def138
Genetic & Molecular Studies
-
Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15(1):70-73. doi:10.1038/ng0197-70
-
Chamberlain SJ, Lalande M. Angelman syndrome, a genomic imprinting disorder of the brain. J Neurosci. 2010;30(30):9958-9963. doi:10.1523/JNEUROSCI.1728-10.2010
-
Mertz LG, Christensen R, Vogel I, et al. Angelman syndrome in Denmark: birth incidence, genetic findings, and age at diagnosis. Am J Med Genet A. 2013;161A(9):2197-2203. doi:10.1002/ajmg.a.36058
-
Petersen MB, Brøndum-Nielsen K, Hansen LK, Wulff K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. Am J Med Genet. 1995;60(3):261-262. doi:10.1002/ajmg.1320600317
Seizure Management
-
Thibert RL, Conant KD, Braun EK, et al. Epilepsy in Angelman syndrome: a questionnaire-based assessment of the natural history and current treatment options. Epilepsia. 2009;50(11):2369-2376. doi:10.1111/j.1528-1167.2009.02108.x
-
Laan LA, Renier WO, Arts WF, et al. Evolution of epilepsy and EEG findings in Angelman syndrome. Epilepsia. 1997;38(2):195-199. doi:10.1111/j.1528-1157.1997.tb01097.x
Genotype-Phenotype Correlations
-
Sahoo T, Peters SU, Madduri NS, et al. Microarray-based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlations. J Med Genet. 2006;43(6):512-516. doi:10.1136/jmg.2005.036913
-
Lossie AC, Whitney MM, Amidon D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001;38(12):834-845. doi:10.1136/jmg.38.12.834
Molecular Mechanisms
-
Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin Genet. 2014;7:93-104. doi:10.2147/TACG.S57386
-
Meng L, Person RE, Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet. 2012;21(13):3001-3012. doi:10.1093/hmg/dds130
-
Greer PL, Hanayama R, Bloodgood BL, et al. The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140(5):704-716. doi:10.1016/j.cell.2010.01.026
-
Burette AC, Judson MC, Li AN, et al. Subcellular organization of UBE3A in human cerebral cortex. Mol Autism. 2018;9:54. doi:10.1186/s13229-018-0238-0
Clinical Features & Management
-
Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat. 2008;4(3):577-584. doi:10.2147/ndt.s2749
-
Evangeliou A, Doulioglou V, Haidopoulou K, et al. Ketogenic diet in a patient with Angelman syndrome. Pediatr Int. 2010;52(5):831-834. doi:10.1111/j.1442-200X.2010.03118.x
-
Braam W, Didden R, Smits MG, Curfs LM. Melatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trial. J Child Neurol. 2008;23(6):649-654. doi:10.1177/0883073808314688
Prognosis & Adult Outcomes
- Larson AM, Shinnick JE, Shaaya EA, et al. Angelman syndrome in adulthood. Am J Med Genet A. 2015;167A(2):331-344. doi:10.1002/ajmg.a.36864
Emerging Therapies
-
Meng L, Ward AJ, Chun S, et al. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518(7539):409-412. doi:10.1038/nature13975
-
Margolis SS, Sell GL, Zbber MA, Bird LM. Angelman syndrome. Neurotherapeutics. 2015;12(3):641-650. doi:10.1007/s13311-015-0361-y
Further Resources
Patient Support Organisations:
- Angelman Syndrome Foundation (US): www.angelman.org
- ASSERT (UK Angelman support): www.angelmanuk.org
- Foundation for Angelman Syndrome Therapeutics (FAST): www.cureangelman.org
- Angelman Syndrome Association Australia: www.angelmansyndrome.org.au
- Unique (Rare Chromosome Disorder Support Group): www.rarechromo.org
Research Registries:
- Global Angelman Syndrome Registry: www.angelmansyndromeregistry.org
- LADDER (Longitudinal Angelman Database and Data Exchange Registry)
Medical Disclaimer: MedVellum content is for educational purposes and clinical reference. Clinical decisions should account for individual patient circumstances. Always consult appropriate specialists. This content does not constitute medical advice for individual patients.
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Genomic Imprinting
- Chromosome 15 Disorders
Differentials
Competing diagnoses and look-alikes to compare.
- Prader-Willi Syndrome
- Rett Syndrome
- Mowat-Wilson Syndrome
Consequences
Complications and downstream problems to keep in mind.
- Refractory Epilepsy
- Developmental Disability