Dermatomyositis
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterised by the combination of proximal muscle weakness... MRCP exam preparation.
What matters first
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterised by the combination of proximal muscle weakness... MRCP exam preparation.
Rapidly progressive ILD (anti-MDA5)
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Rapidly progressive ILD (anti-MDA5)
- Dysphagia (aspiration risk)
- Respiratory muscle weakness
- Malignancy association
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- Polymyositis
- Systemic Lupus Erythematosus
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Dermatomyositis
1. Overview
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterised by the combination of proximal muscle weakness and distinctive cutaneous manifestations. It represents one of the major subgroups of inflammatory myopathies, distinguished from polymyositis and inclusion body myositis by its pathognomonic skin findings and unique pathophysiology. [1,2]
The condition affects both children and adults, with bimodal age distribution. Adult-onset dermatomyositis carries significant implications due to its well-established association with underlying malignancy, occurring in 15-30% of cases. The discovery of myositis-specific autoantibodies has revolutionised understanding of disease phenotypes, with specific antibodies predicting clinical features, complications, and prognosis. [3,4]
Dermatomyositis is now recognised as a complement-mediated microangiopathy affecting muscle, skin, and potentially other organs including the lungs and heart. Early recognition is critical, as prompt immunosuppressive therapy can prevent irreversible muscle damage, and systematic malignancy screening may detect cancers at earlier, more treatable stages. The advent of antibody testing and improved imaging has enhanced diagnostic accuracy and prognostic stratification. [5,6]
2. Epidemiology
Incidence and Prevalence
Dermatomyositis is a rare condition with an estimated incidence of 5-10 cases per million population per year in European and North American populations. [7] The prevalence ranges from 10-20 per million, though geographic and ethnic variations exist. Higher rates have been reported in African American populations compared to Caucasians. [8]
| Statistic | Value | Source |
|---|---|---|
| Annual incidence | 5-10 per million | [7] |
| Prevalence | 10-20 per million | [8] |
| Female to male ratio | 2:1 | [7,8] |
| Malignancy association (adults) | 15-30% | [9,10] |
| ILD prevalence | 30-40% | [11] |
| 5-year mortality | 10-30% | [12] |
Age Distribution
Dermatomyositis exhibits a bimodal age distribution with two distinct peaks. The first peak occurs in childhood between ages 5-15 years (juvenile dermatomyositis), while the second peak affects adults between ages 40-60 years. [7,8] Adult-onset disease carries the significant concern of malignancy association, whereas juvenile disease is more commonly associated with calcinosis and has a lower malignancy risk.
Sex Distribution
There is a clear female predominance with a female to male ratio of approximately 2:1 in adult dermatomyositis. This female preponderance is consistent with other autoimmune conditions and suggests hormonal or genetic factors in disease susceptibility. [7,8]
Risk Factors
Unlike many autoimmune conditions, specific environmental triggers or genetic predispositions for dermatomyositis remain poorly defined. Associations have been reported with:
- HLA-DRB1 alleles (particularly HLA-DRB1*03:01) in Caucasian populations [13]
- UV radiation exposure (may trigger or exacerbate cutaneous manifestations)
- Certain medications (statins, hydroxyurea, though rare)
- Infections (viral triggers proposed but not definitively proven)
- Silica exposure (weak association in some studies)
The strongest association remains with underlying malignancy, particularly in older adults and those with specific antibody profiles.
3. Aetiology and Pathophysiology
Aetiology
The precise aetiology of dermatomyositis remains unknown, but evidence supports an autoimmune process triggered by environmental factors in genetically susceptible individuals. Unlike polymyositis, where T-cell-mediated muscle fibre destruction predominates, dermatomyositis is characterised by complement-mediated microangiopathy. [1,2]
Proposed Triggers:
- Viral infections (influenza, coxsackievirus, HIV)
- Malignancy-associated antigens triggering immune response
- UV radiation (for cutaneous manifestations)
- Certain medications (checkpoint inhibitors, statins)
Pathophysiology
Complement-Mediated Microangiopathy
The fundamental pathophysiological process in dermatomyositis involves activation of the complement cascade with deposition of the membrane attack complex (MAC, C5b-9) on capillary endothelium in muscle and skin. This leads to:
- Capillary destruction: Complement-mediated lysis of endomysial capillaries
- Ischaemia: Reduced capillary density causing muscle ischaemia
- Perifascicular atrophy: Characteristic pattern of muscle fibre atrophy in watershed zones
- Inflammatory infiltrates: Secondary B-cell and T-cell infiltration in perivascular and perimysial regions
Interferon Signature
Type I interferon pathway activation is a hallmark of dermatomyositis pathogenesis. Elevated expression of interferon-inducible genes has been demonstrated in muscle and skin tissue, correlating with disease activity. This interferon signature distinguishes dermatomyositis from other inflammatory myopathies and provides therapeutic targets. [14,15]
Myositis-Specific Autoantibodies
Several myositis-specific autoantibodies (MSAs) have been identified, each associated with distinct clinical phenotypes:
Anti-Mi-2 (10-15% of DM patients)
- Target: Nuclear helicase protein
- Phenotype: Classic dermatomyositis with prominent cutaneous features
- Prognosis: Excellent response to treatment, low malignancy risk
- Mechanism: Unknown pathogenic role
Anti-TIF1γ (Transcriptional Intermediary Factor 1-gamma, 15-20% of adult DM)
- Target: Nuclear protein involved in TGF-β signalling
- Phenotype: Classic dermatomyositis
- Association: Strong association with malignancy (40-70% in adults)
- Clinical utility: Mandates comprehensive malignancy screening
Anti-MDA5 (Melanoma Differentiation-Associated protein 5, 10-30% depending on ethnicity)
- Target: Cytoplasmic viral RNA sensor
- Phenotype: Amyopathic or hypomyopathic dermatomyositis
- Association: Rapidly progressive interstitial lung disease (RP-ILD)
- Prognosis: High mortality if RP-ILD develops (30-50% mortality at 6 months)
- More common in Asian populations
Anti-NXP2 (Nuclear Matrix Protein 2, 15-25% of juvenile DM)
- Target: Nuclear matrix protein
- Phenotype: Severe muscle disease, calcinosis
- Association: Malignancy in adult cases, peripheral oedema
- Prognosis: More resistant to treatment
Anti-SAE (Small Ubiquitin-like Modifier Activating Enzyme)
- Target: Enzymes in SUMO pathway
- Phenotype: Skin-predominant disease, dysphagia
- Association: Low malignancy risk
[3,4,14]
Muscle Pathology
Histopathological examination reveals characteristic features:
- Perifascicular atrophy (pathognomonic finding)
- Perivascular and perimysial inflammatory infiltrates (B-cells, CD4+ T-cells, plasmacytoid dendritic cells)
- Capillary loss and endothelial damage
- MAC deposition on capillaries (demonstrated by C5b-9 immunostaining)
- Regenerating fibres
- Variable necrosis (less prominent than polymyositis)
Skin Pathology
Cutaneous biopsies show:
- Interface dermatitis with vacuolar changes at dermoepidermal junction
- Lymphocytic infiltration
- Mucin deposition in dermis
- MAC deposition on dermal vessels
Malignancy Association
The association between dermatomyositis and malignancy is well-established and clinically significant. The standardised incidence ratio for cancer in dermatomyositis patients is approximately 3-6 times that of the general population. [9,10]
Key Facts:
- Malignancy detected in 15-30% of adult dermatomyositis cases
- Risk highest in first 3-5 years after DM diagnosis
- Bidirectional association: cancer may precede or follow DM diagnosis
- Most cancers are within 1 year before or after DM diagnosis
Most Common Associated Malignancies:
- Ovarian cancer (highest association)
- Lung cancer
- Colorectal cancer
- Gastric cancer
- Nasopharyngeal cancer (particularly in Asian populations)
- Breast cancer
- Non-Hodgkin lymphoma
High-Risk Features for Malignancy:
- Age > 40 years
- Male sex
- Absence of interstitial lung disease
- Cutaneous necrosis or ulceration
- Anti-TIF1γ antibody positivity
- Rapid disease onset
- Elevated CRP (unusual in uncomplicated DM)
[9,10]
4. Clinical Presentation
Cutaneous Features
The skin manifestations of dermatomyositis are often the presenting feature and may precede muscle symptoms by weeks to months. Some patients have skin-predominant disease (amyopathic dermatomyositis) with minimal or no muscle involvement.
Pathognomonic Cutaneous Signs:
Gottron Papules
- Erythematous to violaceous papules or plaques overlying the metacarpophalangeal (MCP) and interphalangeal (IP) joints
- May extend to extensor surfaces of elbows, knees, and ankles
- Often scaly with hyperkeratosis
- May ulcerate in severe cases
- Pathognomonic when present (highly specific)
Gottron Sign
- Erythematous macules over extensor surfaces of joints (without papule formation)
- Less specific than Gottron papules
Heliotrope Rash
- Violaceous to erythematous discolouration of periorbital skin
- Often accompanied by periorbital oedema
- May be subtle and require careful examination
- Named after heliotrope flower colour
- Highly specific when present
Characteristic but Non-Pathognomonic Features:
Shawl Sign
- Photosensitive erythema over upper back, shoulders, and posterior neck
- May extend in V-distribution
- Often indicates active disease
V-Sign
- Erythema in V-distribution over anterior chest and neck
- Photodistributed
Mechanic's Hands
- Hyperkeratotic, cracked, fissured skin on palmar and lateral aspects of fingers
- Resembles manual labourer's hands
- Associated with anti-synthetase antibodies (Jo-1), suggesting overlap syndrome
- May indicate higher risk of ILD
Additional Cutaneous Features:
- Poikiloderma (atrophic skin with hyper/hypopigmentation and telangiectasia)
- Calcinosis cutis (subcutaneous calcium deposits, more common in juvenile DM)
- Periungual changes (dilated capillary loops, cuticular overgrowth)
- Scalp involvement (pruritic erythema, scaling, alopecia)
- Panniculitis (rare)
- Cutaneous ulceration (poor prognostic sign, suggests vasculopathy)
Muscle Features
Proximal Muscle Weakness
The classic presentation involves symmetrical proximal muscle weakness affecting:
- Shoulder girdle: Difficulty lifting arms above head, combing hair, reaching overhead
- Pelvic girdle: Difficulty rising from chair, climbing stairs, getting out of car
The weakness typically develops subacutely over weeks to months, distinguishing it from more acute presentations like viral myositis or rapid presentations of polymyositis.
Functional Limitations:
- Difficulty with activities of daily living requiring arm elevation
- Reduced mobility and falls risk
- Neck flexor weakness (may cause difficulty lifting head from pillow)
- Respiratory muscle weakness (diaphragm, intercostals) in severe cases
Important Characteristics:
- Distal muscles typically spared initially
- Reflexes usually preserved (unless severe atrophy)
- Sensation intact (distinguishes from neuropathies)
- Muscle pain (myalgia) present in 30-50% but not universal
- Muscle tenderness on palpation variable
Dysphagia and Oropharyngeal Involvement
Dysphagia occurs in 30-60% of dermatomyositis patients and results from involvement of pharyngeal and upper oesophageal striated muscles. [16]
Clinical Features:
- Difficulty initiating swallowing
- Nasal regurgitation
- Aspiration risk (potentially life-threatening)
- Dysphonia (vocal cord involvement)
- Oesophageal dysmotility affecting upper two-thirds
Significance:
- Marker of disease severity
- Aspiration pneumonia risk
- Nutritional impairment
- Requires specific assessment and management
Systemic and Extramuscular Manifestations
Interstitial Lung Disease (ILD)
ILD represents one of the most significant complications, occurring in 30-40% of dermatomyositis patients. [11] It is the leading cause of death in anti-MDA5-positive patients.
Patterns:
- Non-specific interstitial pneumonia (NSIP) - most common
- Organising pneumonia (OP/COP)
- Usual interstitial pneumonia (UIP) - worse prognosis
- Diffuse alveolar damage (in rapidly progressive cases)
Clinical Presentation:
- Progressive dyspnoea on exertion
- Dry cough
- Bibasilar inspiratory crackles
- May be asymptomatic (detected on screening)
Anti-MDA5-Associated RP-ILD:
- Rapidly progressive course over weeks
- Extensive ground-glass opacities on CT
- Often amyopathic (minimal muscle disease)
- High mortality (30-50% at 6 months despite aggressive treatment)
- More common in Asian populations
- Requires urgent aggressive immunosuppression
Cardiac Involvement
Cardiac manifestations occur in 10-30% of cases but are often subclinical. [17]
Manifestations:
- Myocarditis (acute or chronic)
- Arrhythmias (conduction defects, atrial/ventricular arrhythmias)
- Congestive heart failure
- Pericarditis (rare)
- Coronary vasculitis (rare)
Detection:
- ECG abnormalities (non-specific ST-T changes, arrhythmias)
- Elevated troponin or BNP
- Echocardiographic abnormalities
- Cardiac MRI (T2-weighted imaging showing oedema)
Arthropathy
Joint involvement occurs in 25-50% of patients:
- Symmetrical polyarthritis resembling rheumatoid arthritis
- Usually non-erosive
- Affects small and large joints
- More common with anti-synthetase antibodies (overlap syndrome)
Constitutional Symptoms
- Fatigue (very common, often debilitating)
- Low-grade fever
- Weight loss
- Raynaud phenomenon (especially in overlap syndromes)
Amyopathic Dermatomyositis
A subset of patients (10-20%) present with characteristic cutaneous features but no clinical muscle weakness despite at least 6 months of observation. Subclinical muscle involvement may be detected on MRI or biopsy. This phenotype is strongly associated with anti-MDA5 antibodies and carries risk of rapidly progressive ILD. [18]
5. Differential Diagnosis
Inflammatory Myopathies
Polymyositis
- Proximal muscle weakness without characteristic DM rash
- T-cell-mediated muscle fibre destruction (different pathophysiology)
- Endomysial inflammatory infiltrates on biopsy
- No perifascicular atrophy
- No capillary MAC deposition
- Diagnosis increasingly questioned (may represent unrecognised IBM or overlap syndromes)
Inclusion Body Myositis (IBM)
- Age > 50 years typically
- Insidious onset (slower progression)
- Distal and proximal weakness (finger flexors, knee extensors)
- Asymmetric weakness common
- Dysphagia frequent
- Poor response to immunosuppression
- Biopsy shows rimmed vacuoles and inclusions
Immune-Mediated Necrotising Myopathy (IMNM)
- Prominent muscle necrosis on biopsy
- Minimal inflammation
- Associated with anti-SRP or anti-HMGCR antibodies
- Statin-associated in some cases
- Severe weakness with very high CK (often > 10,000 U/L)
Overlap Syndromes and Connective Tissue Diseases
Anti-Synthetase Syndrome
- Myositis + ILD + arthritis + mechanic's hands + Raynaud + fever
- Anti-Jo-1 (most common) or other anti-synthetase antibodies
- ILD often more prominent than muscle disease
- Distinct clinical syndrome requiring specific recognition
Systemic Lupus Erythematosus (SLE)
- Malar rash, photosensitivity, mucosal ulcers
- Arthritis, serositis, renal involvement
- Positive ANA, anti-dsDNA, low complement
- Myalgia common but true myositis less frequent
Systemic Sclerosis (Scleroderma)
- Skin thickening and tightness
- Raynaud phenomenon (almost universal)
- Digital ulcers, calcinosis
- ILD common
- Myopathy can occur in overlap syndromes
Mixed Connective Tissue Disease (MCTD)
- Features of SLE, SSc, and myositis
- Anti-U1-RNP antibodies
- Raynaud, puffy hands, arthritis
Muscular Dystrophies
Age-appropriate consideration of genetic muscle diseases:
- Limb-girdle muscular dystrophies
- Facioscapulohumeral dystrophy
- Dysferlinopathy
Distinguishing Features:
- Family history of muscle disease
- Earlier onset (often childhood/adolescence)
- Slowly progressive or static course
- Specific distribution patterns (e.g., facial weakness in FSHD)
- No inflammatory markers or rash
- Biopsy shows dystrophic rather than inflammatory changes
- Genetic testing confirmatory
Drug-Induced Myopathies
Statin-Induced Myopathy
- Exposure to statins
- Usually resolves with drug cessation
- May trigger immune-mediated necrotising myopathy (anti-HMGCR) requiring immunosuppression
- No rash
Checkpoint Inhibitor-Induced Myositis
- Exposure to pembrolizumab, nivolumab, ipilimumab
- Can be severe with cardiac involvement
- Often concurrent myasthenia gravis
- Requires immunosuppression
Other Drugs:
- Hydroxychloroquine (vacuolar myopathy)
- Colchicine (neuromyopathy)
- Alcohol (acute alcoholic myopathy)
Infectious Myositis
Viral Myositis
- Influenza, HIV, Coxsackie virus
- Acute onset
- Self-limiting course usually
- Specific viral testing
Pyomyositis
- Bacterial infection (usually Staphylococcus aureus)
- Localised muscle abscess
- Fever, localised pain and swelling
- MRI shows fluid collection
- More common in tropical regions or immunocompromised
Endocrine and Metabolic Myopathies
Hypothyroid Myopathy
- Proximal weakness, muscle cramps, myalgia
- Elevated CK
- TSH elevated, T4 low
- Resolves with thyroid replacement
Vitamin D Deficiency
- Proximal myopathy, bone pain
- Low 25-OH vitamin D
- Common and often overlooked
Cushing Syndrome
- Steroid-induced myopathy (including exogenous steroids)
- Proximal weakness without CK elevation
- Cushingoid features
Other Considerations
Malignancy-Related
- Paraneoplastic syndrome distinct from DM-associated cancer
- Lambert-Eaton myasthenic syndrome (fatiguable weakness)
- Cancer cachexia
Critical Illness Myopathy
- ICU setting, prolonged immobilisation
- Neuromuscular blocking agents, corticosteroids
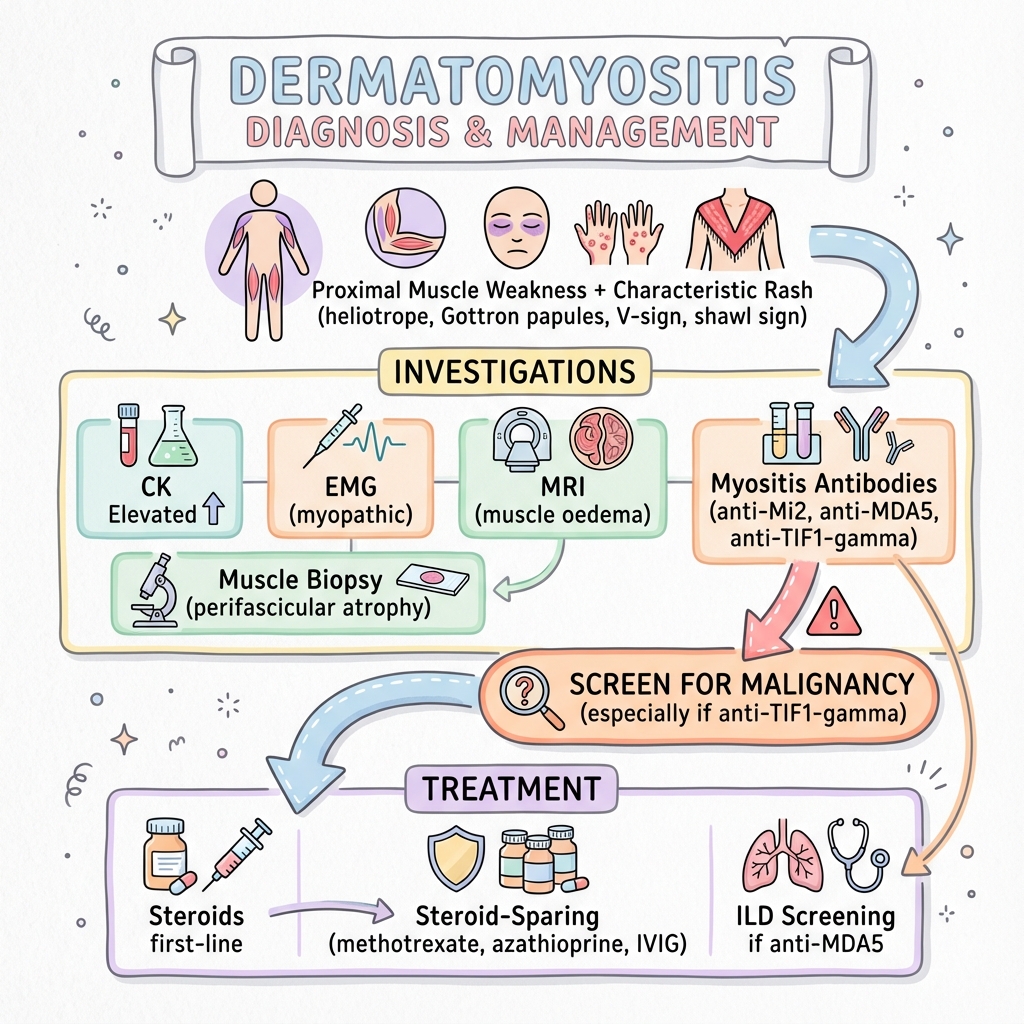
6. Investigations
Muscle Enzymes
Creatine Kinase (CK)
- Elevated in 70-90% of patients with active muscle disease
- Degree of elevation variable (100-10,000+ U/L)
- May be normal in amyopathic DM or chronic/burnt-out disease
- Useful for monitoring disease activity and treatment response
- Less elevated than in immune-mediated necrotising myopathy
Other Muscle Enzymes:
- Aldolase (may be elevated when CK normal)
- LDH (lactate dehydrogenase)
- AST and ALT (often elevated, can be confused with hepatic origin)
Electromyography (EMG)
Needle EMG demonstrates myopathic changes:
- Short-duration, low-amplitude, polyphasic motor unit potentials
- Early recruitment
- Spontaneous fibrillations and positive sharp waves (indicate active muscle damage)
- Distribution: proximal > distal muscles
Utility:
- Confirms myopathic process
- Guides biopsy site selection (abnormal but not end-stage muscle)
- Differentiates from neuropathic disorders
- Less commonly used now with availability of MRI
Magnetic Resonance Imaging (MRI)
MRI has become increasingly valuable in dermatomyositis evaluation. [19]
T2-Weighted and STIR Sequences:
- High signal intensity indicating muscle oedema/inflammation
- Identifies active disease
- Guides biopsy location
- Monitors treatment response
- More sensitive than clinical examination for subclinical involvement
Patterns:
- Patchy or diffuse signal abnormalities
- Predominant proximal muscle involvement
- Perifascicular pattern (specific for DM)
- Subcutaneous oedema
Advantages over Biopsy:
- Non-invasive
- Surveys entire muscle groups
- Distinguishes active inflammation from chronic fibrosis
- Can detect improvement with therapy
Muscle Biopsy
Muscle biopsy remains the gold standard for definitive diagnosis, providing histopathological confirmation and excluding alternative diagnoses.
Characteristic Histological Features:
Perifascicular Atrophy (Pathognomonic)
- 2-10 layers of atrophic fibres at periphery of fascicles
- Results from watershed ischaemia due to capillary loss
- Specific for dermatomyositis (not seen in polymyositis or IBM)
Perivascular and Perimysial Inflammation
- B-lymphocytes, CD4+ T-cells, plasmacytoid dendritic cells
- Predominantly perivascular location
- Perimysial distribution (between fascicles)
- Contrast with endomysial infiltrates in polymyositis
Capillary Changes
- Reduced capillary density (capillary dropout)
- Endothelial hyperplasia
- MAC (C5b-9) deposition on capillaries (immunohistochemistry)
- Diagnostic specificity
Additional Features:
- Variable muscle fibre necrosis and regeneration
- Type 2 fibre atrophy
- Increased connective tissue (chronic cases)
- Alkaline phosphatase positivity at perifascicular fibres
Biopsy Technique:
- Open surgical biopsy preferred (adequate tissue)
- Select muscle: clinically weak but not end-stage (MRI guidance)
- Avoid muscles recently sampled by EMG (artefactual changes)
- Common sites: deltoid, quadriceps, biceps brachii
Autoantibody Testing
Comprehensive myositis antibody panel testing is now standard in suspected dermatomyositis. [3,4]
Myositis-Specific Antibodies (MSAs):
| Antibody | Frequency in DM | Clinical Association | Implications |
|---|---|---|---|
| Anti-Mi-2 | 10-15% | Classic DM, prominent cutaneous features | Good prognosis, excellent treatment response, low malignancy risk |
| Anti-TIF1γ | 15-20% (adults) | Classic DM | High malignancy risk (40-70% in adults), mandates comprehensive cancer screening |
| Anti-MDA5 | 10-30% (ethnic variation) | Amyopathic/hypomyopathic DM | Rapidly progressive ILD risk, high mortality if RP-ILD develops, higher in Asian populations |
| Anti-NXP2 | 15-25% (juvenile), 5-10% (adult) | Severe muscle disease, calcinosis | Malignancy association in adults, treatment-resistant |
| Anti-SAE | Rare (5%) | Skin-predominant disease, dysphagia | Low malignancy risk |
Myositis-Associated Antibodies (MAAs):
- Anti-Ro52 (often concurrent with MSAs, associated with ILD)
- Anti-PM/Scl (overlap with systemic sclerosis)
- Anti-Ku (overlap with systemic sclerosis/SLE)
Anti-Synthetase Antibodies (suggest overlap syndrome):
- Anti-Jo-1 (most common, 20-30% of myositis overall, less common in pure DM)
- Anti-PL-7, Anti-PL-12, Anti-EJ, Anti-OJ, Anti-KS
Routine Autoantibodies:
- ANA (antinuclear antibody): positive in 50-80%, usually speckled or nucleolar pattern
- RF (rheumatoid factor): occasionally positive
- Anti-dsDNA: negative (helps exclude SLE)
Pulmonary Function Testing
Essential screening given 30-40% ILD prevalence. [11]
Tests:
- Spirometry: restrictive pattern (reduced FVC)
- Lung volumes: reduced TLC, RV
- Gas transfer: reduced DLCO (most sensitive early marker)
Indications for HRCT Chest:
- Abnormal PFTs
- Respiratory symptoms
- Anti-MDA5 or anti-synthetase antibodies (high ILD risk)
- Consider screening all DM patients
HRCT Patterns:
- Ground-glass opacities
- Reticular abnormalities
- Consolidation (organising pneumonia)
- Subpleural distribution
- Basal predominance typically
Malignancy Screening
Comprehensive age-appropriate cancer screening is mandatory in adult dermatomyositis. [9,10]
Standard Screening (All Adult DM Patients):
- Detailed history and physical examination
- CT chest/abdomen/pelvis
- Age and sex-appropriate cancer screening:
- Mammography (women > 40)
- Cervical smear (women)
- Colonoscopy (adults > 50 or earlier with symptoms)
- PSA (controversial, discuss with patient)
- Testicular examination
- Baseline and serial CA-125 (ovarian cancer - highest association)
- Nasopharyngoscopy in Asian populations
- Faecal occult blood testing
Enhanced Screening in High-Risk:
- Anti-TIF1γ positive patients
- Age > 60 years
- Rapid onset DM
- Skin necrosis or ulceration
- Elevated CRP (unusual in uncomplicated DM)
Consideration of PET-CT:
- High-risk cases
- Negative initial screening with high suspicion
- Detects occult malignancies
Surveillance:
- Repeat comprehensive screening at 6 months, 12 months, then annually for 3-5 years
- Highest cancer detection within 3 years of DM diagnosis
Cardiac Assessment
Screening Investigations:
- ECG: arrhythmias, conduction defects, non-specific ST-T changes
- Echocardiography: systolic/diastolic dysfunction, regional wall motion abnormalities
- Troponin: may be elevated in myocarditis
- BNP/NT-proBNP: heart failure marker
Advanced Imaging (if cardiac involvement suspected):
- Cardiac MRI: gold standard for myocarditis detection
- "T2-weighted: oedema"
- "Late gadolinium enhancement: fibrosis/necrosis"
- "T1 mapping: diffuse fibrosis"
Other Laboratory Investigations
Inflammatory Markers:
- ESR: often normal or mildly elevated in uncomplicated DM
- CRP: typically normal (elevation suggests infection, malignancy, or overlap syndrome)
Organ Function:
- Renal function: baseline before immunosuppression
- Liver function: monitor for drug toxicity
- Bone profile: calcium, phosphate, vitamin D
7. Diagnostic Criteria
2017 EULAR/ACR Classification Criteria
The 2017 EULAR/ACR classification criteria for idiopathic inflammatory myopathies provide a probability-based scoring system for dermatomyositis. [2]
Scoring Domains:
- Age of symptom onset
- Muscle weakness pattern
- Skin manifestations
- Laboratory findings (muscle enzymes)
- Muscle biopsy features
- Other clinical features
Dermatomyositis-Specific Features (High Weights):
- Heliotrope rash
- Gottron papules
- Gottron sign
Probable DM: Score ≥5.5 without biopsy, ≥6.7 with biopsy Definite DM: Score ≥7.5 without biopsy, ≥8.7 with biopsy
Bohan and Peter Criteria (Historical)
The Bohan and Peter criteria (1975) were historically used but are less specific. [20]
Criteria:
- Symmetrical proximal muscle weakness
- Elevated serum muscle enzymes
- Myopathic changes on EMG
- Characteristic muscle biopsy abnormalities
- Typical skin rash (for DM)
Classification:
- Definite DM: Criterion 5 + any 3 of criteria 1-4
- Probable DM: Criterion 5 + any 2 of criteria 1-4
8. Management
The management of dermatomyositis requires a multidisciplinary approach involving rheumatologists, dermatologists, respiratory physicians, oncologists, and rehabilitation specialists. Treatment aims to suppress immune-mediated inflammation, prevent complications, screen for malignancy, and optimise functional outcomes.
General Principles
Treatment Goals:
- Suppress muscle and skin inflammation
- Prevent irreversible muscle damage and atrophy
- Screen for and treat associated malignancy
- Manage extramuscular manifestations (ILD, cardiac)
- Minimise corticosteroid exposure
- Maintain/restore functional capacity
- Prevent complications
Monitoring Disease Activity:
- Clinical assessment: muscle strength (manual muscle testing, MMT-8)
- Functional measures: getting out of chair, climbing stairs, lifting arms
- CK levels (trend more important than absolute value)
- Global disease activity scores
- MRI for objective assessment
Pharmacological Management
First-Line Immunosuppression
High-Dose Corticosteroids
Corticosteroids remain the cornerstone of initial therapy for dermatomyositis.
Regimen:
- Prednisolone 1 mg/kg/day (maximum 80-100 mg) orally
- Continue high dose for 4-8 weeks until clinical improvement and CK declining
- Gradual taper over 6-12 months to minimum effective dose (typically 5-10 mg/day maintenance)
- Rapid tapering risks disease flare
Severe Cases (respiratory muscle weakness, dysphagia, severe ILD):
- IV methylprednisolone 1 g daily for 3-5 days (pulse therapy)
- Followed by oral high-dose predniso lone
Side Effects Requiring Prophylaxis:
- Osteoporosis: bisphosphonates + calcium/vitamin D
- Gastric protection: PPI
- Hyperglycaemia: monitoring, antidiabetic agents if needed
- Infection prophylaxis: consider PCP prophylaxis (cotrimoxazole) if high dose > 3 months
- Cataracts, glaucoma: ophthalmology review
Steroid-Sparing Agents
Steroid-sparing immunosuppressants should be initiated early (often with or shortly after corticosteroids) to facilitate steroid tapering and prevent long-term steroid toxicity.
Methotrexate
- First-line steroid-sparing agent
- Dose: 15-25 mg weekly (oral or subcutaneous)
- Onset of action: 6-12 weeks
- Efficacy: effective for muscle and skin disease
- Monitoring: FBC, renal/liver function every 4-8 weeks
- Contraindications: pregnancy, significant renal/liver impairment, ILD (relative contraindication)
- Folate supplementation: 5 mg weekly (separate day from MTX)
Azathioprine
- Alternative first-line agent
- Dose: 2-3 mg/kg/day (maximum 200 mg)
- Onset: 8-12 weeks
- Check TPMT levels before starting (guides dosing, toxicity risk)
- Monitoring: FBC, liver function every 4-8 weeks
- Well-tolerated, safe in pregnancy (Category D)
Mycophenolate Mofetil
- Increasingly used as first-line or second-line
- Dose: 2-3 g daily in divided doses
- Efficacy: particularly for ILD-associated disease
- Monitoring: FBC every 4-8 weeks
- Contraindication: pregnancy
Calcineurin Inhibitors (Tacrolimus, Cyclosporine)
- Effective for refractory muscle and skin disease
- Particularly effective for anti-MDA5-associated DM and ILD
- Tacrolimus: 2-4 mg daily (target trough 5-10 ng/mL)
- Monitoring: renal function, drug levels, blood pressure
- Side effects: nephrotoxicity, hypertension, diabetes
Intravenous Immunoglobulin (IVIG)
IVIG is highly effective for refractory dermatomyositis, particularly for cutaneous and muscle manifestations.
Indications:
- Refractory muscle weakness despite conventional immunosuppression
- Refractory skin disease (especially pruritus, ulceration)
- Dysphagia (particularly effective)
- Patients unable to tolerate conventional immunosuppressants
- Pregnancy (safe)
Regimen:
- 2 g/kg divided over 2-5 days per month
- Response within 2-4 infusions typically
- Long-term maintenance often required
Evidence:
- RCT evidence supports efficacy
- Well-tolerated
- Expensive, access may be limited
Biological Therapies
Rituximab (Anti-CD20)
- Most evidence-based biological therapy for myositis
- Indications: refractory disease despite conventional treatment
- Regimen: 1 g IV at weeks 0 and 2, repeat at 6 months
- Alternative: 375 mg/m² weekly for 4 weeks
- Efficacy: effective in 80-90% of refractory cases
- Particularly effective for anti-synthetase syndrome
- Monitor: immunoglobulin levels, infusion reactions, PML risk (extremely rare)
Other Biologics (Less Evidence):
- Anti-TNF agents (infliximab, adalimumab): limited efficacy, may worsen disease
- Abatacept (CTLA4-Ig): case series show promise
- Tocilizumab (anti-IL-6): case reports
- JAK inhibitors (tofacitinib, baricitinib): emerging evidence, particularly for refractory skin disease
Management of Rapidly Progressive ILD (Anti-MDA5)
Anti-MDA5-positive dermatomyositis with rapidly progressive ILD requires urgent aggressive immunosuppression.
Regimen:
- High-dose IV methylprednisolone 1 g daily × 3-5 days
- Followed by oral prednisolone 1 mg/kg
- Plus dual or triple immunosuppression:
- Tacrolimus (target trough 5-10 ng/mL)
- Mycophenolate mofetil 2-3 g daily OR cyclophosphamide (500-750 mg/m² monthly)
- Consider IVIG
- Rituximab in refractory cases
Monitoring:
- Serial PFTs and HRCT
- Clinical deterioration despite treatment common
- High mortality (30-50% at 6 months)
Cutaneous Disease Management
Topical Therapies:
- High-potency corticosteroids for skin lesions
- Tacrolimus ointment for facial rash
Photoprotection:
- Broad-spectrum sunscreen (SPF 50+)
- Protective clothing, avoid peak sun hours
- UV radiation exacerbates cutaneous disease
Systemic Therapy for Refractory Skin Disease:
- Hydroxychloroquine 200-400 mg daily (first-line for skin)
- Methotrexate
- IVIG (particularly effective for pruritus)
- JAK inhibitors (tofacitinib): emerging evidence
Malignancy Management
Treatment of underlying malignancy is essential in malignancy-associated dermatomyositis.
Principles:
- Definitive cancer treatment per oncology protocols
- Dermatomyositis may improve with successful cancer treatment
- Continue immunosuppression for DM during cancer therapy (coordination with oncology)
- Complete cancer remission may lead to DM remission in some cases
Rehabilitation and Supportive Care
Physiotherapy:
- Early mobilisation (avoid bed rest)
- Graduated exercise program
- Isometric exercises during acute phase
- Progressive resistance training during recovery
- Prevents contractures and improves functional outcomes
Occupational Therapy:
- Assessment of activities of daily living
- Assistive devices
- Energy conservation techniques
Swallowing Assessment (if dysphagia present):
- Speech and language therapy evaluation
- Modified diet consistency
- Swallowing strategies
- Consider enteral feeding if severe aspiration risk
Respiratory Physiotherapy:
- Breathing exercises
- Secretion clearance if ILD
- Monitor respiratory muscle strength
Management of Specific Complications
Calcinosis:
- Difficult to treat
- Options: diltiazem, bisphosphonates, IVIG, surgical excision if symptomatic
Cardiac Involvement:
- Myocarditis: high-dose corticosteroids, immunosuppression
- Arrhythmias: cardiology referral, antiarrhythmic agents, pacemaker if indicated
- Heart failure: standard HF management
9. Complications
| Complication | Frequency | Clinical Features | Management |
|---|---|---|---|
| Interstitial Lung Disease | 30-40% | Progressive dyspnoea, dry cough, crackles, restrictive PFTs, HRCT changes | Mycophenolate, tacrolimus, cyclophosphamide, aggressive immunosuppression for RP-ILD |
| Malignancy | 15-30% (adults) | Variable depending on cancer type | Comprehensive screening, definitive cancer treatment |
| Aspiration Pneumonia | 10-20% (with dysphagia) | Fever, cough, chest X-ray consolidation | Antibiotics, swallowing assessment, modified diet, consider NG/PEG feeding |
| Respiratory Muscle Weakness | 5-10% severe | Dyspnoea, orthopnoea, Type 2 respiratory failure | High-dose immunosuppression, non-invasive ventilation, ICU if respiratory failure |
| Cardiac Involvement | 10-30% | Arrhythmias, heart failure, myocarditis | Immunosuppression, cardiology referral, device therapy if needed |
| Calcinosis | 40% (juvenile), 10% (adult) | Subcutaneous calcium deposits, pain, ulceration, functional impairment | Difficult to treat; diltiazem, bisphosphonates, surgical excision |
| Infection | Variable | Opportunistic infections (PCP, herpes zoster) due to immunosuppression | PCP prophylaxis, vaccination (pre-immunosuppression), prompt treatment |
| Steroid-Induced Complications | Almost universal with prolonged use | Osteoporosis, diabetes, cataracts, avascular necrosis | Bone protection, screening, steroid-sparing agents |
Mortality
Overall 5-year mortality ranges from 10-30% depending on series and era. [12] Major causes of death include:
- Rapidly progressive ILD (especially anti-MDA5)
- Malignancy
- Respiratory failure
- Cardiac complications
- Infection (often iatrogenic from immunosuppression)
Favourable Prognostic Factors:
- Younger age
- Anti-Mi-2 positivity
- Isolated skin or muscle involvement
- Good response to initial therapy
Poor Prognostic Factors:
- Anti-MDA5 with RP-ILD
- Anti-TIF1γ with malignancy
- Older age
- Significant ILD
- Cardiac involvement
- Delay in diagnosis and treatment
10. Prognosis
With modern immunosuppressive therapy, the majority of dermatomyositis patients achieve disease control and functional improvement. However, prognosis varies significantly based on clinical phenotype, antibody profile, and complications.
Overall Outcomes:
- 60-80% achieve good disease control with treatment
- 10-20% have refractory disease requiring multiple agents
- Functional recovery possible but may be incomplete
- Many patients require long-term low-dose maintenance therapy
- Relapses common with rapid steroid tapering
Quality of Life:
- Fatigue often persistent even with disease control
- Chronic pain in subset of patients
- Skin manifestations may be disfiguring
- Functional limitations improve but may not normalise
Long-Term Monitoring:
- Regular follow-up for disease activity assessment
- Surveillance for malignancy (first 3-5 years)
- Monitoring for treatment toxicity
- Bone density scans (corticosteroid-induced osteoporosis)
- Cardiovascular risk management
11. Prevention and Screening
Primary Prevention
No specific primary prevention strategies exist given unclear aetiology.
General Measures:
- Photoprotection (may reduce cutaneous flares)
- Smoking cessation (general health, reduces ILD risk)
- Avoid unnecessary medications that may trigger myositis
Screening
At-Risk Populations: No specific high-risk populations identified for screening, as genetic predisposition is weak.
Malignancy Screening: As detailed in Investigations section, comprehensive age-appropriate cancer screening is mandatory for all adult dermatomyositis patients at diagnosis and during follow-up.
12. Key Guidelines
2017 EULAR/ACR Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies
- Lundberg IE et al., Ann Rheum Dis 2017 [2]
- Provides validated classification criteria
- Facilitates uniform patient selection for trials and studies
EULAR Recommendations for Management of Adult Dermatomyositis and Polymyositis
- Not yet published as comprehensive guideline
- Based on expert consensus and evolving evidence
British Society for Rheumatology Guidelines
- Recommends early aggressive immunosuppression
- Steroid-sparing agents to minimize steroid toxicity
- Multidisciplinary approach
13. Examination Focus
Common Exam Questions
Written Examination Questions:
-
"A 55-year-old woman presents with 3-month history of proximal muscle weakness and a violaceous rash around her eyes. CK is elevated. What is the most likely diagnosis and what is your initial management?"
-
"What are the indications for malignancy screening in dermatomyositis? Which malignancies are most commonly associated?"
-
"Describe the histopathological features that distinguish dermatomyositis from polymyositis on muscle biopsy."
-
"A patient with newly diagnosed dermatomyositis is found to have anti-MDA5 antibodies. What are the clinical implications and how would you manage this patient?"
-
"List the characteristic cutaneous features of dermatomyositis. Which are pathognomonic?"
Clinical Examination Scenarios:
- "Examine this patient's hands"
- Looking for Gottron papules, mechanic's hands, nail fold changes
- "This patient has muscle weakness. Perform a focused neurological examination"
- Looking for proximal weakness, preserved reflexes and sensation
- "Examine this patient's skin"
- Looking for heliotrope rash, shawl sign, V-sign, Gottron papules
Viva Topics:
- Myositis-specific antibodies and their clinical associations
- Differential diagnosis of proximal myopathy
- Management of rapidly progressive ILD in anti-MDA5-positive DM
- Malignancy screening protocol in dermatomyositis
- Distinguishing dermatomyositis from polymyositis (clinical, histological, immunological)
Viva Points
Opening Statement:
"Dermatomyositis is an idiopathic inflammatory myopathy characterised by proximal muscle weakness and distinctive cutaneous manifestations, most notably Gottron papules and heliotrope rash. It affects approximately 5-10 per million population annually with a female predominance and bimodal age distribution affecting children and adults aged 40-60."
Key Facts to Mention:
Pathophysiology:
- Complement-mediated microangiopathy affecting endomysial capillaries
- Type I interferon signature
- Perifascicular atrophy on muscle biopsy (pathognomonic)
Clinical Features:
- Gottron papules (pathognomonic): erythematous plaques over MCP/IP joints
- Heliotrope rash: violaceous periorbital discoloration
- Proximal muscle weakness: shoulder and hip girdles
- Dysphagia in 30-60%
Myositis-Specific Antibodies:
- Anti-Mi-2: classic DM, good prognosis
- Anti-TIF1γ: high malignancy association (40-70% in adults)
- Anti-MDA5: rapidly progressive ILD, high mortality
- Anti-NXP2: calcinosis, severe disease
Investigations:
- CK elevated (70-90%)
- EMG: myopathic changes
- MRI: muscle edema, guides biopsy
- Muscle biopsy: perifascicular atrophy, perimysial inflammation, MAC deposition
- Myositis antibody panel
- Malignancy screening: mandatory in adults
Management:
- High-dose corticosteroids: prednisolone 1 mg/kg/day
- Steroid-sparing agents: methotrexate, azathioprine (initiated early)
- IVIG for refractory disease
- Rituximab for refractory cases
- Aggressive immunosuppression for RP-ILD (tacrolimus, mycophenolate, cyclophosphamide)
Complications:
- ILD (30-40%), rapidly progressive in anti-MDA5
- Malignancy (15-30% adults)
- Aspiration pneumonia
- Cardiac involvement
Prognosis:
- 60-80% achieve disease control
- 5-year mortality 10-30%
- Anti-Mi-2: excellent prognosis
- Anti-MDA5 with RP-ILD: 30-50% mortality at 6 months
Common Mistakes
❌ Mistakes That Fail Candidates:
-
Missing malignancy screening: Failing to recognise mandatory comprehensive cancer screening in adult dermatomyositis, especially with anti-TIF1γ positivity
-
Confusing DM with polymyositis: Not recognising that skin findings are essential for DM diagnosis; polymyositis has no rash
-
Not recognising anti-MDA5 implications: Missing the urgency of rapidly progressive ILD in anti-MDA5-positive patients requiring immediate aggressive immunosuppression
-
Inadequate dysphagia assessment: Underestimating aspiration risk in patients with dysphagia; this is potentially life-threatening
-
Monotherapy with steroids alone: Not initiating steroid-sparing agents early, leading to steroid toxicity
-
Missing cardiac involvement: Failing to screen for cardiac complications which are a significant cause of mortality
-
Describing polymyositis biopsy findings for DM: Confusing endomysial inflammation (polymyositis) with perimysial/perivascular inflammation and perifascicular atrophy (DM)
-
Wrong antibody associations: Confusing antibody-phenotype associations (e.g., stating anti-Jo-1 is common in DM when it's more associated with anti-synthetase syndrome)
Model Answers
Q: Describe your approach to a patient presenting with proximal muscle weakness and a rash suggestive of dermatomyositis.
Model Answer:
"I would approach this systematically with three parallel aims: confirming the diagnosis, assessing disease severity and complications, and screening for malignancy.
Initial Assessment: First, I would take a detailed history focusing on the onset and progression of muscle weakness, functional limitations such as difficulty climbing stairs or lifting objects, any dysphagia or respiratory symptoms, and constitutional symptoms. I would ask about cutaneous features and photograph the rash for documentation. A drug history is essential to exclude drug-induced myopathy, and I would inquire about symptoms suggesting malignancy.
Examination: I would perform a full examination including careful assessment of characteristic skin lesions - looking specifically for Gottron papules over the MCP and IP joints, heliotrope rash around the eyes, shawl sign, V-sign, and mechanic's hands. Muscle examination would assess proximal muscle groups, looking for symmetrical weakness of shoulder and hip girdles, while checking that reflexes and sensation are preserved. I would assess for dysphagia, respiratory muscle weakness, and examine for signs of ILD with chest auscultation.
Investigations: Blood tests would include CK (elevated in 70-90%), muscle enzymes (aldolase, LDH, AST/ALT), inflammatory markers (ESR usually normal, CRP elevation suggests complication), renal and liver function, and a comprehensive myositis antibody panel including anti-Mi-2, anti-TIF1γ, anti-MDA5, anti-NXP2, and anti-synthetase antibodies.
MRI of affected muscle groups would demonstrate oedema on T2/STIR sequences and guide biopsy site selection. Muscle biopsy would be performed to confirm diagnosis and demonstrate perifascicular atrophy, perimysial inflammation, and MAC deposition on capillaries.
Screening for complications would include pulmonary function tests with DLCO, HRCT chest if PFTs abnormal or high ILD risk (anti-MDA5, anti-synthetase), ECG and echocardiography, and swallowing assessment if dysphagia present.
Malignancy Screening: Given the 15-30% malignancy association in adults, I would perform comprehensive age-appropriate cancer screening including CT chest/abdomen/pelvis, age-specific screening (mammography, colonoscopy, cervical smear), CA-125 for ovarian cancer, and consider PET-CT in high-risk cases particularly with anti-TIF1γ positivity.
Management: I would initiate high-dose corticosteroids with prednisolone 1 mg/kg/day along with bone protection, PPI, and monitoring for hyperglycaemia. Concurrently, I would start a steroid-sparing agent such as methotrexate 15-25 mg weekly with folic acid, or azathioprine 2 mg/kg/day after checking TPMT levels. For refractory disease, I would consider IVIG or rituximab.
If anti-MDA5 antibodies are positive, I would urgently assess for ILD and if rapidly progressive ILD is present, initiate aggressive triple immunosuppression with IV methylprednisolone, tacrolimus, and mycophenolate or cyclophosphamide.
I would arrange multidisciplinary input including rheumatology, dermatology, respiratory medicine if ILD present, and physiotherapy for rehabilitation. Regular monitoring of disease activity through clinical assessment, CK levels, and functional measures would guide treatment adjustments. Surveillance for malignancy would continue for 3-5 years with repeat screening at 6 months, 12 months, and annually."
14. References
-
Dalakas MC. Inflammatory Muscle Diseases. N Engl J Med. 2015;372(18):1734-1747. doi:10.1056/NEJMra1402225
-
Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76(12):1955-1964. doi:10.1136/annrheumdis-2017-211468
-
Satoh M, Tanaka S, Ceribelli A, Calise SJ, Chan EK. A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin Rev Allergy Immunol. 2017;52(1):1-19. doi:10.1007/s12016-015-8510-y
-
Mecoli CA, Albayda J, Tiniakou E, et al. Myositis autoantibodies: A clinical perspective. Curr Opin Rheumatol. 2020;32(6):612-619. doi:10.1097/BOR.0000000000000745
-
Ladislau L, Suarez-Calvet X, Toquet S, et al. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain. 2018;141(6):1609-1621. doi:10.1093/brain/awy105
-
Opinc AH, Makowska JS. Antisynthetase syndrome - much more than just a myopathy. Semin Arthritis Rheum. 2021;51(1):72-83. doi:10.1016/j.semarthrit.2020.09.020
-
Bendewald MJ, Wetter DA, Li X, Davis MD. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146(1):26-30. doi:10.1001/archdermatol.2009.328
-
Meyer A, Meyer N, Schaeffer M, et al. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford). 2015;54(1):50-63. doi:10.1093/rheumatology/keu289
-
Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100. doi:10.1016/S0140-6736(00)03540-6
-
Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum. 2013;65(11):2954-2962. doi:10.1002/art.38093
-
Bonella F, Costabel U. Interstitial lung disease in idiopathic inflammatory myopathies. Curr Opin Pulm Med. 2014;20(5):445-451. doi:10.1097/MCP.0000000000000084
-
Marie I, Hatron PY, Dominique S, et al. Short-term and long-term outcomes of interstitial lung disease in polymyositis and dermatomyositis: a series of 107 patients. Arthritis Rheum. 2011;63(11):3439-3447. doi:10.1002/art.30513
-
Rothwell S, Cooper RG, Lundberg IE, et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis. 2016;75(8):1558-1566. doi:10.1136/annrheumdis-2015-208119
-
Ladislau L, Suarez-Calvet X, Toquet S, et al. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain. 2018;141(6):1609-1621. doi:10.1093/brain/awy105
-
Galindo-Feria AS, Albrecht I, Fernandes-Cerqueira C, et al. Proinflammatory Histidyl-Transfer RNA Synthetase-Specific CD4+ T Cells in the Blood and Lungs of Patients With Idiopathic Inflammatory Myopathies. Arthritis Rheumatol. 2020;72(1):179-191. doi:10.1002/art.41075
-
Marie I, Menard JF, Hachulla E, et al. Infectious complications in polymyositis and dermatomyositis: a series of 279 patients. Semin Arthritis Rheum. 2011;41(1):48-60. doi:10.1016/j.semarthrit.2010.08.003
-
Diederichsen LP, Simonsen JA, Diederichsen AC, Hvidsten S, Hougaard M, Junker P, Davidsen ML, Hansen V, Iversen LV, Hilberg O. Cardiac abnormalities in adult patients with polymyositis or dermatomyositis as assessed by noninvasive modalities. Arthritis Care Res (Hoboken). 2016;68(7):1012-1020. doi:10.1002/acr.22772
-
Gerami P, Schope JM, McDonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54(4):597-613. doi:10.1016/j.jaad.2005.10.041
-
Pipitone N, Notarnicola A, Levrì L, Morra E, Salvarani C. Do we really need muscle biopsy in suspected idiopathic inflammatory myopathies? Clin Exp Rheumatol. 2021;39(5):1153-1161.
-
Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344-347. doi:10.1056/NEJM197502132920706
Last Updated: 2026-01-05
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Autoimmune Disease Fundamentals
Differentials
Competing diagnoses and look-alikes to compare.
- Polymyositis
- Systemic Lupus Erythematosus
- Systemic Sclerosis
- Inclusion Body Myositis
Consequences
Complications and downstream problems to keep in mind.
- Interstitial Lung Disease
- Malignancy Screening