Hereditary Haemochromatosis
Hereditary haemochromatosis (HH) is an autosomal recessive disorder of iron metabolism characterized by excessive intest... MRCP exam preparation.
What matters first
Hereditary haemochromatosis (HH) is an autosomal recessive disorder of iron metabolism characterized by excessive intest... MRCP exam preparation.
Cirrhosis with elevated ferritin
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Cirrhosis with elevated ferritin
- Hepatocellular carcinoma in cirrhotic patient
- Dilated cardiomyopathy with diabetes
- Diabetes with hepatomegaly and bronze skin
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- Secondary Iron Overload
- Porphyria Cutanea Tarda
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Hereditary Haemochromatosis
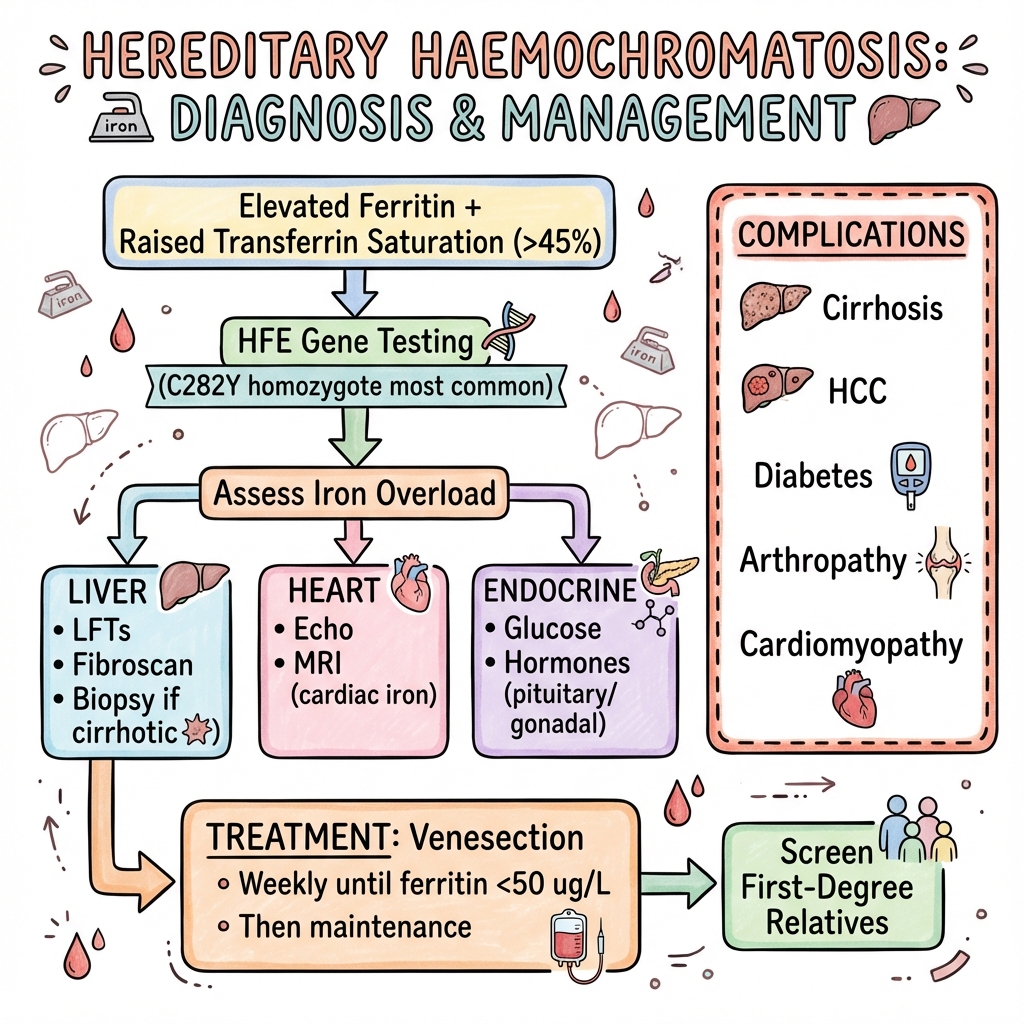
1. Overview
Hereditary haemochromatosis (HH) is an autosomal recessive disorder of iron metabolism characterized by excessive intestinal iron absorption leading to progressive iron accumulation in parenchymal tissues. It represents the most common inherited metabolic disorder in populations of Northern European ancestry. The condition results from mutations in genes regulating iron homeostasis, most commonly the HFE gene located on chromosome 6. [1,2]
Without treatment, excess iron deposits in the liver, pancreas, heart, pituitary gland, joints, and skin, causing multiorgan dysfunction. The classic triad of "bronze diabetes" comprises cirrhosis, diabetes mellitus, and skin hyperpigmentation. However, this presentation is now uncommon due to earlier diagnosis through biochemical screening. The hallmark laboratory findings are elevated transferrin saturation (typically > 45%) and elevated serum ferritin, with confirmation by HFE genotyping demonstrating C282Y homozygosity in the majority of cases. [3,4]
Early diagnosis and treatment with therapeutic phlebotomy (venesection) can prevent organ damage and normalize life expectancy. However, once cirrhosis develops, the risk of hepatocellular carcinoma remains significantly elevated even after iron depletion. First-degree relatives of affected individuals require screening given the autosomal recessive inheritance pattern. [5,6]
2. Epidemiology
Hereditary haemochromatosis demonstrates marked ethnic variation, with highest prevalence in populations of Northern European descent, particularly those of Celtic ancestry. The condition is significantly rarer in Asian, African, and indigenous populations. [7]
| Epidemiological Feature | Value | Population | Reference |
|---|---|---|---|
| HFE C282Y homozygote frequency | 1:200-300 | Northern European | [1,7] |
| HFE C282Y heterozygote frequency | 1:8-10 | Northern European | [7] |
| Clinical penetrance (C282Y homozygotes) | 1-10% males, 0.5-2% females | General population | [8,9] |
| Biochemical penetrance | 40-70% males, 10-30% females | C282Y homozygotes | [8] |
| Male:female clinical expression | 3-5:1 | Affected individuals | [2] |
| Peak age of presentation (males) | 40-60 years | Clinical cohorts | [2,10] |
| Peak age of presentation (females) | 50-70 years (post-menopause) | Clinical cohorts | [2,10] |
| Prevalence of cirrhosis at diagnosis | 5-10% | Contemporary cohorts | [11] |
| Lifetime HCC risk (with cirrhosis) | 20-30% | Cirrhotic patients | [12] |
Geographic and Ethnic Distribution
The C282Y mutation arose approximately 60-70 generations ago in Northern Europe, likely in a Celtic ancestor, explaining the high carrier frequency in Irish, Scottish, Welsh, and Scandinavian populations. Ireland has the highest prevalence, with C282Y homozygosity affecting approximately 1:83 individuals. [7]
In contrast, C282Y is rare in sub-Saharan African, East Asian, and indigenous Australian populations (frequency less than 1:10,000). Other HFE mutations, including H63D and S65C, are more geographically distributed but cause clinically significant iron overload only in specific compound heterozygous states. [1,7]
Incomplete Penetrance
A critical epidemiological feature is the incomplete penetrance of HFE-related haemochromatosis. While C282Y homozygosity is relatively common (1:200-300 in Northern Europeans), only a minority develop clinically significant disease requiring treatment. Population studies demonstrate that approximately 40-70% of male C282Y homozygotes and 10-30% of female homozygotes develop biochemical iron overload (elevated transferrin saturation and ferritin). [8,9]
The proportion progressing to symptomatic disease requiring therapeutic phlebotomy is even lower, estimated at 1-10% of male homozygotes and 0.5-2% of female homozygotes. This phenotypic variability reflects modifying genetic factors, dietary iron intake, blood loss (menstruation, pregnancy, blood donation), alcohol consumption, and concurrent liver disease. [8,9]
Sex Differences
Men develop clinical manifestations 2-3 decades earlier than women and are 3-5 times more likely to present with symptomatic disease. This sex difference reflects protective factors in premenopausal women, including menstrual blood loss (approximately 25-30 mg iron monthly) and pregnancy-related iron demands. After menopause, women progressively accumulate iron and may develop clinical manifestations if untreated. [2,10]
Temporal Trends
Earlier detection through increased awareness and biochemical screening has shifted the presentation pattern. While historical cohorts frequently presented with advanced cirrhosis and the classic "bronze diabetes" triad, contemporary patients are often identified at asymptomatic or minimally symptomatic stages through screening of first-degree relatives or incidental laboratory findings. This earlier diagnosis has substantially reduced the prevalence of advanced organ damage at presentation. [11]
3. Aetiology and Pathophysiology
Genetic Basis
Hereditary haemochromatosis results from mutations in genes regulating iron homeostasis, predominantly the HFE gene located on chromosome 6p21.3. [1,2]
HFE Mutations:
-
C282Y (c.845G>A): The predominant mutation causing classical HFE-associated haemochromatosis, accounting for 80-85% of cases in Northern European populations. This missense mutation (cysteine→tyrosine at position 282) disrupts a disulfide bond critical for protein folding, preventing normal HFE-β2-microglobulin complex formation and cell surface expression. [1,13]
-
H63D (c.187C>G): A milder variant with reduced penetrance. H63D homozygosity rarely causes significant iron overload. Compound heterozygosity (C282Y/H63D) accounts for approximately 5% of clinical haemochromatosis but typically with less severe phenotype than C282Y homozygosity. [1,13]
-
S65C (c.193A>T): A rare variant. Compound heterozygosity with C282Y (C282Y/S65C) may cause mild iron overload in some individuals. [1]
Non-HFE Haemochromatosis (Rare):
While HFE mutations account for > 90% of hereditary haemochromatosis in European populations, non-HFE forms exist: [14]
- Type 2A (juvenile haemochromatosis): HJV gene (hemojuvelin) mutations - autosomal recessive, presents age less than 30 with severe iron overload, cardiomyopathy, and hypogonadism
- Type 2B: HAMP gene (hepcidin) mutations - similar severe early-onset phenotype
- Type 3: TFR2 gene (transferrin receptor 2) mutations - intermediate severity
- Type 4: SLC40A1 gene (ferroportin) mutations - autosomal dominant, distinct pattern with elevated ferritin but normal/low transferrin saturation
Molecular Pathophysiology
Hepcidin-Ferroportin Axis:
Normal iron homeostasis is regulated by hepcidin, a 25-amino acid peptide hormone synthesized by hepatocytes. Hepcidin binds to ferroportin (the only known cellular iron exporter) on duodenal enterocytes, hepatocytes, and macrophages, triggering ferroportin internalization and degradation. This reduces iron export from cells into plasma. [15]
HFE Protein Function:
Wild-type HFE protein forms a complex with transferrin receptor 1 (TFR1) and β2-microglobulin at the hepatocyte cell surface. When transferrin-bound iron binds to TFR1, the HFE-β2-microglobulin complex dissociates and interacts with transferrin receptor 2 (TFR2), initiating signaling cascades that upregulate hepcidin transcription via the BMP-SMAD pathway (particularly BMP6). [15,16]
C282Y Mutation Consequence:
The C282Y mutation prevents proper HFE-β2-microglobulin complex formation and cell surface expression. Mutant HFE protein is retained in the endoplasmic reticulum and degraded. This disrupts the iron-sensing mechanism, resulting in inappropriately low hepcidin production despite iron overload. [15,16]
Low Hepcidin State:
Reduced hepcidin levels fail to downregulate ferroportin, leading to:
- Increased duodenal iron absorption: Ferroportin remains expressed on basolateral enterocyte membranes, allowing excessive iron transfer from enterocytes to plasma (2-3x normal absorption rate)
- Increased macrophage iron release: Ferroportin on macrophages continues recycling iron from senescent erythrocytes into circulation
- Decreased hepatocyte iron retention: Ferroportin remains functional, preventing compensatory iron storage in hepatocytes
The net result is progressive accumulation of iron in plasma and subsequent non-transferrin-bound iron (NTBI) formation when transferrin becomes saturated. [15,16]
Tissue Iron Deposition:
NTBI is readily taken up by parenchymal cells (hepatocytes, pancreatic β-cells, cardiomyocytes) via non-specific mechanisms, particularly ZIP14 transporters. This leads to progressive parenchymal iron loading while reticuloendothelial macrophages remain relatively iron-depleted (reversed pattern compared to secondary iron overload). [15]
Oxidative Damage:
Free intracellular iron catalyzes Fenton reactions (Fe²⁺ + H₂O₂ → Fe³⁺ + OH· + OH⁻), generating highly reactive hydroxyl radicals. These cause: [17]
- Lipid peroxidation of cell membranes
- DNA strand breaks and mutations
- Protein carbonylation and dysfunction
- Mitochondrial damage
- Activation of hepatic stellate cells → fibrogenesis
Fibrosis Progression:
Chronic iron-mediated oxidative stress activates hepatic stellate cells, which transform into collagen-producing myofibroblasts. Progressive extracellular matrix deposition leads to bridging fibrosis and eventual cirrhosis. Cirrhosis typically requires total body iron stores > 20 g (normal 3-4 g). [17]
Carcinogenesis:
Iron-catalyzed oxidative DNA damage promotes mutagenesis. Additionally, iron dysregulates cell cycle checkpoints and enhances inflammation-mediated carcinogenesis. The hepatocellular carcinoma risk in cirrhotic haemochromatosis patients is 20-200 fold elevated compared to the general population, and persists even after iron depletion. [12,18]
Summary of Pathophysiological Cascade
- HFE gene mutation (typically C282Y homozygosity)
- Defective HFE protein → impaired iron sensing
- Inappropriately low hepcidin synthesis
- Increased ferroportin activity → enhanced intestinal iron absorption
- Progressive iron accumulation in plasma (transferrin saturation > 45%, then NTBI formation)
- Parenchymal cell iron uptake (liver, pancreas, heart, pituitary, joints)
- Intracellular iron-catalyzed oxidative damage
- Organ dysfunction: hepatic fibrosis→cirrhosis→HCC, cardiomyopathy, diabetes, arthropathy, hypogonadism
4. Clinical Presentation
The clinical manifestations of haemochromatosis result from progressive iron deposition in multiple organs. Presentation varies from asymptomatic biochemical abnormalities detected on screening to advanced multiorgan dysfunction.
Temporal Evolution
- Asymptomatic phase: Elevated transferrin saturation and ferritin without symptoms (often detected on screening or incidental testing)
- Non-specific symptoms: Fatigue, lethargy, arthralgias (frequently dismissed or misattributed)
- Organ-specific manifestations: Develop with progressive iron accumulation (typically total body iron > 10-20 g)
- Advanced "bronze diabetes": Now rare due to earlier detection
Symptoms
| Symptom | Frequency | Clinical Context | Reference |
|---|---|---|---|
| Fatigue, lethargy | 60-75% | Most common presenting symptom; often dismissed as non-specific | [2,10] |
| Arthralgia | 40-50% | Particularly metacarpophalangeal joints (2nd/3rd); progressive | [2,10] |
| Abdominal pain | 20-30% | Hepatomegaly-related discomfort | [10] |
| Loss of libido | 15-30% | Secondary to hypogonadism (gonadotropin deficiency) | [10] |
| Erectile dysfunction | 20-40% (males) | Hypogonadotropic hypogonadism | [10] |
| Amenorrhea | Variable (premenopausal) | Pituitary iron deposition | [10] |
| Dyspnea, reduced exercise tolerance | 15-25% | Cardiac involvement | [19] |
| Polyuria, polydipsia | Variable | Diabetes mellitus development | [2] |
Signs
General Examination
Skin Changes:
- Bronze or slate-grey hyperpigmentation (60-70% of symptomatic patients)
- Results from combined melanin and iron deposition in dermis
- Most prominent in sun-exposed areas, flexures, scars, genitalia
- Often subtle and overlooked [2,10]
Organ-Specific Signs
Hepatic:
- Hepatomegaly (75-95% at presentation with symptoms; often smooth, non-tender)
- Stigmata of chronic liver disease (in advanced cirrhosis):
- Spider naevi
- Palmar erythema
- Gynecomastia
- Testicular atrophy
- Ascites (decompensated cirrhosis)
- Splenomegaly (portal hypertension) [2,11]
Cardiac:
- Arrhythmias (supraventricular and ventricular)
- Signs of congestive heart failure:
- Elevated jugular venous pressure
- Peripheral edema
- Bibasal crackles
- Displaced apex beat (dilated cardiomyopathy)
- Restrictive cardiomyopathy pattern (particularly in juvenile haemochromatosis) [19]
Endocrine:
- Testicular atrophy (hypogonadism)
- Loss of secondary sexual hair
- Thin, dry skin (hypothyroidism - rare complication)
- Acanthosis nigricans (insulin resistance) [2]
Musculoskeletal:
- Arthropathy:
- Metacarpophalangeal (MCP) joint involvement (2nd/3rd MCP most characteristic)
- Proximal interphalangeal joints
- Wrists, knees, hips, shoulders (progressive disease)
- Bony swelling, restricted movement
- Chondrocalcinosis on X-ray (calcium pyrophosphate deposition) [20]
Other:
- Absence of reticuloendothelial iron loading (unlike secondary iron overload - spleen typically not enlarged unless portal hypertension present)
Classic Triad: "Bronze Diabetes"
Historically the hallmark presentation, now uncommon due to earlier diagnosis:
- Bronze/grey skin pigmentation: Melanin + iron deposition
- Diabetes mellitus: Pancreatic β-cell iron damage (typically insulin-requiring)
- Cirrhosis: Advanced hepatic fibrosis with complications
This triad indicates advanced disease with total body iron stores typically > 20 g. [2,10]
Atypical Presentations
- Isolated arthropathy (occasionally precedes other manifestations by years)
- Dilated cardiomyopathy or arrhythmia in young adult (consider juvenile haemochromatosis if age less than 30)
- Unexplained chronic transaminitis
- Incidental ferritin elevation on routine testing
- Family screening of asymptomatic C282Y homozygote
Age and Sex Differences
Males:
- Typically present age 40-60 years
- More likely to have advanced organ damage
- Higher likelihood of cirrhosis at diagnosis [2,10]
Females:
- Typically present age 50-70 years (post-menopause)
- Protected by menstruation and pregnancy during reproductive years
- Lower overall penetrance [2,10]
5. Differential Diagnosis
When evaluating patients with elevated ferritin, iron studies abnormalities, or clinical features suggestive of iron overload, several conditions require consideration.
Primary Differentials
| Condition | Key Distinguishing Features | Transferrin Saturation | Ferritin | Genetics |
|---|---|---|---|---|
| HFE haemochromatosis | Northern European ancestry; C282Y homozygote | > 45% (typically > 60%) | Elevated (often > 1000 μg/L) | C282Y/C282Y (85%); C282Y/H63D (5%) |
| Non-HFE haemochromatosis | Early onset (less than 30 years); severe cardiomyopathy; family history | > 45% | Very high (> 2000 μg/L) | HJV, HAMP, TFR2 mutations |
| Ferroportin disease | Autosomal dominant; ferritin↑↑ but TfSat normal/low | Normal or ↓ | Elevated | SLC40A1 mutation |
| Secondary iron overload | Transfusion history; chronic anemia; alcohol | Variable | Elevated | Normal HFE |
| Porphyria cutanea tarda | Photosensitive blistering rash; urine fluoresces | Normal or mildly ↑ | Mildly-moderately ↑ | UROD mutation; may coexist with HFE |
| Metabolic syndrome | Obesity, insulin resistance, dyslipidemia | Normal | Mildly ↑ (100-500) | Normal HFE |
| Chronic liver disease | Viral hepatitis, NAFLD, alcohol | Normal or ↓ | Mildly-moderately ↑ | Variable |
| Inflammation/infection | CRP elevated; acute illness | Low-normal | Elevated (acute phase reactant) | Normal HFE |
Secondary Causes of Iron Overload
Transfusion-Related:
- Multiple red cell transfusions (each unit ~200-250 mg iron; no physiological excretion mechanism)
- Thalassemia major
- Sickle cell disease with chronic transfusions
- Myelodysplastic syndromes
- Aplastic anemia
- Pattern: Reticuloendothelial (spleen, bone marrow, Kupffer cells) iron loading initially, then parenchymal [1]
Increased Absorption:
- Chronic hemolytic anemias with ineffective erythropoiesis (thalassemia intermedia, congenital dyserythropoietic anemias)
- Mechanism: Low hepcidin secondary to erythropoietic drive despite iron overload
- Sideroblastic anemias
- Chronic liver disease (alcohol, viral hepatitis, NAFLD) - disrupts hepcidin regulation [1]
Dietary/Iatrogenic:
- Excessive oral iron supplementation (usually requires concomitant liver disease to cause significant overload)
- Bantu siderosis (traditional beer brewed in iron pots - now rare)
- Parenteral iron overload (excessive intravenous iron therapy)
Hyperferritinemia Without Iron Overload
Ferritin is an acute phase reactant; many conditions elevate ferritin without true iron excess: [3]
Inflammatory Conditions:
- Acute and chronic infections
- Autoimmune diseases (Still's disease, systemic lupus erythematosus)
- Chronic kidney disease
- Malignancy
Hepatocellular Damage:
- Acute hepatitis (viral, drug-induced, ischemic)
- Chronic liver disease (NAFLD, alcohol, viral hepatitis)
- Ferritin released from damaged hepatocytes
Metabolic Syndrome:
- Insulin resistance
- NAFLD
- Obesity
- Mechanism: Inflammatory state with elevated hepcidin; usually mild ferritin elevation (100-500 μg/L)
Haematological:
- Hemophagocytic lymphohistiocytosis (very high ferritin > 10,000 μg/L)
- Adult-onset Still's disease (ferritin > 3000 μg/L, often > 10,000 μg/L)
Key Discriminators:
- Transferrin saturation typically normal or low in hyperferritinemia without iron overload
- CRP, ESR elevated in inflammatory states
- Liver function tests abnormal in hepatocellular damage
- Absent iron deposition on liver biopsy if performed
Hereditary Hyperferritinemia-Cataract Syndrome
- Autosomal dominant
- L-ferritin gene mutations
- Elevated ferritin (typically 500-2000 μg/L) from childhood
- Normal iron studies (transferrin saturation, serum iron)
- Early-onset cataracts (childhood/adolescence)
- No iron overload; phlebotomy contraindicated [1]
Aceruloplasminemia
- Rare autosomal recessive
- Ceruloplasmin gene mutation
- Iron accumulates in liver, pancreas, retina, basal ganglia
- Anemia, diabetes, retinal degeneration, neurological symptoms (dystonia, ataxia)
- Low serum ceruloplasmin and copper
- Elevated ferritin; normal/low transferrin saturation [1]
6. Investigations
The diagnostic approach combines biochemical iron studies, genetic testing, and assessment of organ iron loading and damage.
Screening and Initial Investigations
Indications for Screening:
- First-degree relatives of patients with HFE haemochromatosis (highest yield)
- Unexplained chronic liver disease or elevated transaminases
- Unexplained diabetes, particularly with liver disease
- Cardiomyopathy or arrhythmias (especially less than 50 years)
- Arthropathy, particularly 2nd/3rd MCP involvement
- Hypogonadism
- Chronic fatigue with hepatomegaly
- Porphyria cutanea tarda [3,4]
First-Line Screening Tests:
| Test | Normal Range | Haemochromatosis Finding | Interpretation | Reference |
|---|---|---|---|---|
| Transferrin saturation | 20-45% | > 45% (fasting) | Best screening test; elevated early; repeat if > 45% | [3,4] |
| > 60% in C282Y homozygotes typically | Suggests iron overload if persistent | |||
| Serum ferritin | Males: 30-300 μg/L | > 200 μg/L (women) | Reflects iron stores but non-specific (acute phase reactant) | [3,4] |
| Females: 15-200 μg/L | > 300 μg/L (men) | |||
| Often > 1000 μg/L in symptomatic HH | Very high levels suggest advanced disease | |||
| Serum iron | 10-30 μmol/L | Elevated | Less useful alone; diurnal variation | [3] |
| Total iron-binding capacity (TIBC) | 45-70 μmol/L | Low-normal or decreased | Transferrin saturation = serum iron/TIBC × 100 | [3] |
Transferrin Saturation as Primary Screening Tool:
Transferrin saturation (TfSat) is superior to ferritin for initial screening because:
- Elevated earlier in disease course (before ferritin rises)
- More specific for iron overload (ferritin elevated by inflammation, liver disease, malignancy)
- Correlates with circulating non-transferrin-bound iron and risk of parenchymal deposition
- Cutoff > 45% has sensitivity ~94% and specificity ~86% for HFE haemochromatosis [3,4]
Important considerations:
- Fasting sample preferred (slight elevation postprandially)
- Repeat if > 45% to confirm persistence
- May be lower in menstruating women despite iron accumulation
- May be transiently low during acute inflammation (hepcidin surge)
Ferritin Interpretation Pitfalls:
- Acute phase reactant: elevated in inflammation, infection, malignancy
- Released from damaged hepatocytes: elevated in acute/chronic liver disease
- Metabolic syndrome: mild elevation (100-500 μg/L) common without iron overload
- Very high levels (> 1000 μg/L) suggest: (1) significant iron overload, (2) acute hepatitis, (3) hemophagocytic syndrome, or (4) malignancy
Recommended Screening Algorithm:
- Measure fasting transferrin saturation and ferritin
- If TfSat > 45% and/or ferritin elevated: repeat after 1-2 weeks
- If persistently abnormal: proceed to HFE genotyping
- Exclude secondary causes (CRP, liver function, alcohol history, medication review)
Confirmatory Genetic Testing
HFE Genotyping:
- Indication: Persistently elevated transferrin saturation (> 45%) and/or ferritin
- Method: PCR-based detection of common HFE mutations
- Mutations tested: C282Y, H63D, S65C
- Sensitivity: ~95% for Northern European haemochromatosis (C282Y homozygotes)
- Interpretation:
- "C282Y/C282Y homozygote: Confirms HFE haemochromatosis diagnosis (80-85% of cases)"
- "C282Y/H63D compound heterozygote: May cause mild-moderate iron overload (~5% of cases)"
- "C282Y heterozygote (C282Y/WT): Not sufficient for HFE haemochromatosis diagnosis; investigate other causes"
- "H63D/H63D homozygote: Rarely causes significant iron overload"
- "Wild-type/wild-type: Consider non-HFE haemochromatosis or secondary iron overload [1,3,4]"
Non-HFE Genetic Testing:
If iron overload confirmed but HFE genotype does not explain phenotype, consider sequencing:
- HJV gene (juvenile haemochromatosis, type 2A)
- HAMP gene (juvenile haemochromatosis, type 2B)
- TFR2 gene (type 3)
- SLC40A1 gene (ferroportin disease, type 4)
- Particularly if: age less than 30 years, severe cardiomyopathy, autosomal dominant pattern, or very high ferritin with normal/low TfSat [14]
Quantification of Iron Stores
Liver MRI (T2 or R2):**
- Non-invasive quantification of hepatic iron concentration
- T2* relaxation time inversely proportional to tissue iron
- R2* (1/T2*) correlates with liver iron concentration (LIC)
- Advantages: Non-invasive, reproducible, suitable for monitoring
- Interpretation:
- "Normal LIC: less than 36 μmol/g dry weight (less than 2 mg/g)"
- "Mild overload: 36-80 μmol/g"
- "Moderate: 80-160 μmol/g"
- "Severe: > 160 μmol/g"
- Useful for: confirming iron overload when ferritin equivocal, monitoring treatment response
- Note: Less accurate if severe fibrosis/cirrhosis present [3,21]
Cardiac MRI (T2):*
- Assesses myocardial iron loading
- Critical in juvenile haemochromatosis where cardiac involvement determines prognosis
- T2* less than 20 ms indicates myocardial iron deposition with risk of arrhythmia/cardiomyopathy
- T2* less than 10 ms indicates severe iron loading with high risk of heart failure
- Indication: Age less than 30 at presentation, cardiac symptoms, very high ferritin (> 2000 μg/L), non-HFE haemochromatosis [19,21]
Liver Biopsy:
- Historically gold standard; now rarely required due to non-invasive alternatives
- Indications: (1) Uncertainty about diagnosis, (2) elevated transaminases suggesting concurrent liver disease, (3) assessment of fibrosis stage, (4) ferritin > 1000 μg/L (higher cirrhosis risk)
- Findings:
- Prussian blue staining demonstrates parenchymal (hepatocyte) iron deposition (grade 3-4)
- Minimal reticuloendothelial (Kupffer cell) iron (unlike secondary overload)
- "Quantitative hepatic iron concentration (HIC): > 80 μmol/g dry weight abnormal; > 160 μmol/g strongly suggests haemochromatosis"
- "Hepatic iron index (HIC/age): > 1.9 previously used (less relevant now with genetic testing)"
- Fibrosis staging (Ishak or METAVIR)
- Risks: Bleeding, pain, sampling error [3,11]
Assessment of Organ Damage
Liver Assessment:
- Liver function tests: ALT, AST, ALP, bilirubin, albumin, INR
- Often normal or mildly elevated
- Moderate elevation (ALT less than 5× ULN typical)
- Suggest concurrent pathology if ALT > 5× ULN (viral hepatitis, NASH, alcohol)
- Abdominal ultrasound: Hepatomegaly, altered echogenicity, focal lesions, splenomegaly (portal hypertension)
- Transient elastography (FibroScan): Non-invasive assessment of liver stiffness (fibrosis surrogate)
- less than 7 kPa: minimal fibrosis
-
12-14 kPa: cirrhosis likely
- Iron deposition may falsely elevate readings
- HCC surveillance (if cirrhosis present): 6-monthly ultrasound + alpha-fetoprotein (AFP) [11,12]
Cardiac Assessment:
- ECG: Arrhythmias (atrial fibrillation, ventricular ectopy, heart block)
- Echocardiography:
- Left ventricular systolic dysfunction (dilated cardiomyopathy pattern)
- Diastolic dysfunction (restrictive pattern in severe cases)
- Reduced ejection fraction
- Atrial enlargement
- Cardiac MRI T2*: If cardiac symptoms, age less than 30, or very high ferritin [19]
Endocrine Assessment:
- Diabetes screening: Fasting glucose, HbA1c, oral glucose tolerance test (OGTT)
- Diabetes present in ~30-60% of symptomatic patients
- Hypogonadism: Testosterone (males), LH, FSH, estradiol (females)
- Hypogonadotropic hypogonadism pattern (low testosterone/estradiol with low-normal LH/FSH)
- Thyroid function: TSH, free T4 (hypothyroidism rare)
- Pituitary function: If clinical features suggest hypopituitarism (comprehensive pituitary hormone panel) [2,10]
Musculoskeletal Assessment:
- X-rays of affected joints:
- MCP 2/3, wrists, knees, hips
- "Findings: joint space narrowing, subchondral sclerosis, osteophytes, cyst formation"
- Chondrocalcinosis (calcium pyrophosphate deposition) - characteristic
- DEXA scan: Osteoporosis risk assessment (hypogonadism, direct iron effect on bone) [20]
Monitoring During Treatment
- Pre-phlebotomy: Hemoglobin and hematocrit (avoid anemia)
- Ferritin: Every 3 months during induction, before each maintenance venesection
- Transferrin saturation: Monitor alongside ferritin
- Liver function tests: 6-12 monthly
- HCC surveillance: If cirrhosis present, continue lifelong (6-monthly ultrasound + AFP) even after iron depletion [3,4,11]
7. Management
The cornerstone of haemochromatosis management is iron depletion through therapeutic phlebotomy (venesection). Early treatment prevents organ damage and normalizes life expectancy; established cirrhosis is irreversible, requiring lifelong HCC surveillance.
Indications for Treatment
Confirmed HFE Haemochromatosis (C282Y Homozygote):
- Ferritin > 200 μg/L (women) or > 300 μg/L (men) with transferrin saturation > 45%
- Evidence of iron overload on MRI or biopsy
- Clinical evidence of iron-related organ damage
Compound Heterozygotes (C282Y/H63D):
- Treat if clinically significant iron overload (ferritin > 500-1000 μg/L and elevated TfSat)
Asymptomatic C282Y Homozygotes:
- If ferritin and TfSat normal/borderline: monitor annually
- Lifestyle advice (avoid iron supplements, vitamin C supplements, excessive alcohol)
- Consider treatment if ferritin rising trend [3,4]
Therapeutic Phlebotomy (Venesection)
Induction Phase:
Goal: Deplete excess iron stores to low-normal range
| Parameter | Protocol | Rationale |
|---|---|---|
| Volume per session | 450-500 mL whole blood | Removes ~200-250 mg iron per session |
| Frequency | Weekly (or twice weekly if tolerated) | Balances efficacy with patient tolerance |
| Target ferritin | 50-100 μg/L (some guidelines less than 50 μg/L) | Indicates iron-replete status without deficiency |
| Duration | Variable: 6 months to 2-3 years | Depends on baseline iron stores |
| Monitoring | Hemoglobin before each venesection | Ensure Hb > 11 g/dL to proceed safely |
| Ferritin every 3 months | Track progress toward target |
Pre-Venesection Checklist:
- Check hemoglobin/hematocrit (do not venesect if Hb less than 11 g/dL or symptoms of anemia)
- Assess patient wellbeing (defer if acute illness)
- Adequate hydration encouraged [3,4]
Induction Phase Endpoints:
- Ferritin 50-100 μg/L (some centers target less than 50 μg/L)
- Transferrin saturation normalizes (typically less than 50%)
- Hemoglobin falls to low-normal or mild anemia (indicator of iron depletion)
- May require 20-150 venesections depending on baseline iron burden [3,4]
Calculation of Total Iron Excess:
Ferritin approximately correlates with total body iron stores:
- Each 1 μg/L ferritin ≈ 8-10 mg total body iron (rough estimate)
- Example: Ferritin 2000 μg/L → ~16,000-20,000 mg excess iron (normal stores ~3000-4000 mg)
- Each 500 mL venesection removes ~250 mg iron
- Estimated venesections required: 16,000 mg / 250 mg = 64 sessions
This is approximate; actual requirement varies with erythropoietic response, diet, and baseline stores.
Physiological Adaptation to Phlebotomy:
Weekly venesection induces erythropoiesis, increasing iron demand. Marrow expansion draws on iron stores to maintain hemoglobin. Over months, ferritin declines progressively. Once stores depleted, further venesection induces mild iron-deficiency anemia (hemoglobin 11-12 g/dL), signaling cessation of induction phase.
Hepcidin Suppression:
Phlebotomy-induced erythropoiesis suppresses hepcidin (erythroferrone mechanism), potentially increasing dietary iron absorption. Despite this, venesection effectively depletes stores due to obligate iron loss.
Maintenance Phase:
Goal: Prevent iron reaccumulation while maintaining hemoglobin
| Parameter | Protocol | Rationale |
|---|---|---|
| Frequency | Every 2-4 months | Individualized based on ferritin trend |
| Volume | 450-500 mL | Standard unit |
| Target ferritin | 50-100 μg/L | Prevent both iron excess and deficiency |
| Monitoring | Ferritin before each session; annual transferrin saturation | Adjust frequency to maintain target |
| Duration | Lifelong | Continued intestinal iron hyperabsorption |
Factors Affecting Maintenance Frequency:
- Dietary iron intake (red meat consumption)
- Continued menstruation (women require less frequent venesection)
- Blood donation (menstruating women)
- Alcohol intake (avoid or minimize - enhances iron absorption and liver damage)
- Vitamin C supplementation (avoid - enhances iron absorption) [3,4]
Alternative Iron Depletion
Iron Chelation Therapy:
Reserved for patients unable to tolerate phlebotomy:
- Severe anemia (e.g., concomitant thalassemia, chronic kidney disease)
- Inadequate venous access
- Severe cardiac disease precluding fluid shifts
- Patient refusal/phobia of venesection [3]
Chelation Agents:
| Agent | Route | Dose | Mechanism | Limitations |
|---|---|---|---|---|
| Deferoxamine | Subcutaneous or IV infusion | 20-40 mg/kg 5-7 times/week | Binds free iron; excreted in urine | Burdensome administration; expensive; auditory/visual toxicity |
| Deferasirox | Oral | 20-30 mg/kg daily | Once-daily tablet | Expensive; GI side effects; renal toxicity; less effective than phlebotomy |
| Deferiprone | Oral | 75-100 mg/kg/day (divided TID) | Alternative oral chelator | Agranulocytosis risk; arthropathy; less data in HH |
Note: Phlebotomy is far superior (faster, cheaper, safer, better tolerated) when feasible. Chelation rarely used in HFE haemochromatosis. [3]
Lifestyle and Dietary Modifications
Avoid:
- Iron supplements: Unless documented iron-deficiency anemia develops (rare)
- Vitamin C supplements: Enhances intestinal iron absorption and mobilizes iron from stores
- Excess alcohol: Increases iron absorption, synergistic hepatotoxicity; limit to less than 2 units/day or abstain if liver disease present
- Raw shellfish: Risk of Vibrio vulnificus septicemia (iron-overloaded patients susceptible to siderophilic organisms)
- Uncooked seafood: Similar risk [3,22]
Dietary Iron:
- Moderate restriction not essential (phlebotomy overcomes dietary intake)
- Avoid extreme high-iron diets (excessive red meat, fortified cereals)
- Tea with meals may reduce iron absorption (tannins bind iron) [22]
Permitted:
- Blood donation (if meets other eligibility criteria) can contribute to iron removal
Management of Organ Complications
Cirrhosis:
- HCC surveillance: 6-monthly abdominal ultrasound + AFP (lifelong, even after iron depletion)
- Variceal screening: Endoscopy if cirrhosis confirmed (portal hypertension assessment)
- Liver transplantation: Consider if decompensated cirrhosis (MELD score, Child-Pugh C)
- Iron depletion improves post-transplant outcomes if achieved pre-transplant [11,12]
Diabetes Mellitus:
- Insulin often required (β-cell destruction may be irreversible)
- Standard diabetes management (lifestyle, metformin, insulin as needed)
- Iron depletion may improve glycemic control in some patients [2,10]
Cardiomyopathy:
- Aggressive iron depletion (weekly or twice-weekly phlebotomy; consider chelation if severe)
- Standard heart failure management (ACE inhibitors, beta-blockers, diuretics, device therapy)
- Cardiac MRI T2* monitoring
- Cardiac function may improve with iron depletion if treated early [19]
Hypogonadism:
- Testosterone replacement therapy (males): transdermal gel or intramuscular injections
- Monitor PSA, hematocrit (testosterone increases erythropoiesis)
- Estrogen/progesterone replacement (females) if symptomatic
- Fertility counseling; pituitary function may not recover despite iron depletion [2,10]
Arthropathy:
- Does not improve with iron depletion (established chondrocalcinosis and cartilage damage irreversible)
- Analgesics (paracetamol, NSAIDs - caution in liver disease)
- Intra-articular corticosteroid injections for acute pseudogout flares
- Arthroplasty for severe joint destruction (hip, knee) [20]
Family Screening
First-Degree Relatives (Parents, Siblings, Children):
Autosomal recessive inheritance; siblings have 25% risk of being C282Y homozygotes.
Recommended Approach:
- HFE genotyping of all first-degree relatives
- Iron studies (transferrin saturation, ferritin) in genotype-positive relatives
- Repeat iron studies every 2-5 years in C282Y homozygotes even if initially normal (penetrance variable) [3,4,5]
Genetic Counseling:
- C282Y homozygote patients should inform relatives
- Offspring of C282Y homozygote: 100% carriers (C282Y heterozygotes) unless partner also carrier
- If partner is C282Y heterozygote (1:8-10 in Northern Europeans), 50% risk of affected homozygous offspring
- Partner screening offered if planning children [5]
Prenatal/Preimplantation Genetic Diagnosis:
Generally not recommended given:
- Incomplete penetrance (most C282Y homozygotes remain asymptomatic or minimally affected)
- Effective treatment available (phlebotomy)
- Normal life expectancy with early treatment [5]
Monitoring and Long-Term Follow-Up
Treated Haemochromatosis Without Cirrhosis:
- Maintenance phlebotomy every 2-4 months (titrated to ferritin 50-100 μg/L)
- Annual liver function tests
- Annual assessment for diabetes (fasting glucose, HbA1c)
- Clinical review annually
- No routine HCC surveillance required (risk very low without cirrhosis) [3,4]
Treated Haemochromatosis With Cirrhosis:
- Maintenance phlebotomy every 2-4 months
- Lifelong HCC surveillance: 6-monthly ultrasound + AFP (even after iron depletion - risk persists)
- 6-monthly liver function tests
- Endoscopic variceal screening as per local protocol (typically every 2-3 years if no varices; annual if varices present)
- Clinical review every 6 months
- Assess for hepatic decompensation (ascites, encephalopathy, variceal bleeding) [11,12]
Special Situations
Pregnancy:
- C282Y homozygous women generally tolerate pregnancy well (iron transferred to fetus)
- Hold venesection during pregnancy (fetal iron demands reduce maternal stores)
- Monitor ferritin and hemoglobin
- Resume phlebotomy postpartum after lactation complete or as needed [3]
Concurrent Liver Disease:
- HFE haemochromatosis + viral hepatitis (HCV, HBV): synergistic fibrosis risk; treat both
- HFE + alcohol: strongly synergistic hepatotoxicity; abstinence mandatory
- HFE + NAFLD: common coexistence; treat iron overload; manage metabolic syndrome
- Phlebotomy may improve transaminases and fibrosis progression [3,11]
Non-HFE Haemochromatosis (Juvenile, Type 2):
- Earlier presentation (age less than 30), more severe iron loading
- Aggressive phlebotomy (twice weekly if tolerated) or combination phlebotomy + chelation
- Cardiac MRI T2* monitoring essential (cardiomyopathy leading cause of death)
- Intensive cardiac iron depletion may reverse cardiomyopathy if caught early [14,19]
8. Complications
Complications arise from progressive iron deposition and oxidative damage to parenchymal organs. Early diagnosis and treatment prevent most complications; established complications (cirrhosis, arthropathy) are often irreversible.
| Complication | Frequency | Pathophysiology | Prevention | Management | Reversibility | Reference |
|---|---|---|---|---|---|---|
| Hepatic fibrosis → cirrhosis | 5-10% at diagnosis (contemporary cohorts) | Iron-catalyzed oxidative stress → stellate cell activation → collagen deposition | Early iron depletion before fibrosis established | Phlebotomy; HCC surveillance; liver transplantation if decompensated | Established cirrhosis irreversible; fibrosis may partially regress if non-cirrhotic | [11,17] |
| Hepatocellular carcinoma (HCC) | 20-30% lifetime risk if cirrhotic; 200-fold increased risk vs general population | Iron-induced oxidative DNA damage + mutagenesis; chronic inflammation | Prevent cirrhosis (early treatment); lifelong surveillance if cirrhotic | Resection, ablation, transplantation, or TACE depending on stage | N/A (cancer) | [12,18] |
| Diabetes mellitus | 30-60% of symptomatic patients | Pancreatic β-cell iron deposition → apoptosis → insulin deficiency | Early iron depletion | Insulin therapy often required; glucose control improves with iron depletion in some | Variable; partial recovery possible if early | [2,10] |
| Dilated cardiomyopathy | 15-30% (higher in juvenile HH) | Myocardial iron → oxidative damage → myocyte dysfunction; diastolic dysfunction → systolic dysfunction | Early iron depletion; aggressive treatment if cardiac T2* low | Intensive phlebotomy/chelation; standard heart failure therapy; pacemaker/ICD if indicated | May improve if treated early (T2* > 10 ms); poor prognosis if severe | [19] |
| Arrhythmias | 20-40% | Iron disrupts cardiac conduction; fibrosis | Early iron depletion | Antiarrhythmics; ablation; pacemaker/ICD | May improve with iron depletion | [19] |
| Hypogonadotropic hypogonadism | 30-50% of males with advanced disease | Pituitary iron deposition → gonadotroph dysfunction → low LH/FSH | Early iron depletion | Testosterone replacement (males); estrogen/progesterone (females) | Usually irreversible despite iron depletion | [2,10] |
| Arthropathy | 40-50% | Iron deposition in synovium → calcium pyrophosphate formation → chondrocalcinosis; cartilage damage | Early iron depletion (may prevent progression but not reverse) | Analgesia; intra-articular steroids for acute flares; arthroplasty if severe | Not reversible; iron depletion does not improve established arthropathy | [20] |
| Skin hyperpigmentation (bronze) | 60-70% of symptomatic patients | Melanin + iron deposition in dermis | Early iron depletion | Iron depletion | Gradually improves (months-years) with iron depletion | [2] |
| Osteoporosis/osteopenia | 25-40% | Hypogonadism; direct iron effect on osteoblasts | Treat hypogonadism; early iron depletion | Calcium/vitamin D; bisphosphonates; testosterone replacement | Partial improvement | [20] |
| Increased infection risk | Variable | Iron promotes growth of siderophilic organisms (Yersinia, Vibrio, Listeria) | Avoid high-risk foods (raw shellfish); iron depletion | Prompt antibiotics; avoid empiric fluoroquinolones (resistance common in Yersinia) | Resolves with iron depletion | [22] |
| Hypothyroidism | less than 5% | Pituitary or thyroid iron deposition | Early iron depletion | Levothyroxine replacement | Variable | [2] |
| Porphyria cutanea tarda | Coexists in ~1-2% (HFE mutation promotes PCT expression) | Reduced UROD activity; iron promotes uroporphyrin accumulation | Iron depletion | Phlebotomy (treats both HH and PCT); hydroxychloroquine | Skin lesions resolve with iron depletion | [3] |
Cirrhosis-Specific Complications
Once cirrhosis develops (5-10% of contemporary cohorts), patients face standard cirrhosis complications:
- Portal hypertension: Varices, splenomegaly, ascites, hepatorenal syndrome
- Hepatic encephalopathy: Ammonia metabolism impaired
- Coagulopathy: Reduced synthetic function (clotting factors)
- Hepatocellular carcinoma: 20-30% lifetime risk; persists despite iron depletion [11,12]
Key Point: HCC risk remains elevated in cirrhotic patients even after complete iron depletion, necessitating lifelong surveillance. Non-cirrhotic patients have very low HCC risk. [12]
Prognostic Factors for Complications
Factors Predicting Adverse Outcomes:
- Age > 40-50 at diagnosis (longer duration of iron accumulation)
- Male sex (earlier, more severe iron loading)
- Ferritin > 1000 μg/L (marker of advanced iron overload and higher cirrhosis risk)
- Established cirrhosis (irreversible; HCC risk persists)
- Hepatomegaly (indicates significant liver involvement)
- Alcohol consumption (synergistic hepatotoxicity)
- Concurrent liver disease (viral hepatitis, NAFLD) [2,10,11]
Protective Factors:
- Diagnosis before age 40
- Treatment before cirrhosis develops
- Female sex (menstruation protective until menopause)
- Ferritin less than 1000 μg/L at diagnosis
- Asymptomatic detection (family screening) [2,10]
9. Prognosis
The prognosis of hereditary haemochromatosis depends critically on the stage at diagnosis and initiation of treatment.
Overall Prognosis
| Clinical Scenario | Life Expectancy | Key Outcomes | Reference |
|---|---|---|---|
| Treated before cirrhosis | Normal | No excess mortality; minimal morbidity if adherent to maintenance phlebotomy | [2,6] |
| Treated with established cirrhosis | Reduced | 5-year survival ~50-60%; HCC risk 20-30% lifetime despite iron depletion | [11,12] |
| Untreated symptomatic disease | Markedly reduced | Progressive multiorgan failure; median survival less than 5 years from symptom onset historically | [2] |
| Asymptomatic C282Y homozygote (normal ferritin) | Normal | Most remain asymptomatic lifelong; annual monitoring recommended | [8,9] |
Impact of Treatment Timing
Early Treatment (Pre-Cirrhotic):
- Complete prevention of cirrhosis, HCC, cardiomyopathy if ferritin normalized before organ damage
- Symptom resolution (fatigue, skin pigmentation improve)
- Normal life expectancy
- Quality of life comparable to general population [2,6]
Late Treatment (Post-Cirrhotic):
- Cirrhosis irreversible (fibrosis may partially regress but architectural distortion persists)
- HCC risk 20-30% lifetime; persists despite complete iron depletion
- Hepatic decompensation risk (ascites, variceal bleeding, encephalopathy)
- Reduced life expectancy (5-year survival ~50-60% with cirrhosis)
- Liver transplantation may be required [11,12]
Specific Complication Outcomes
Hepatocellular Carcinoma:
- Annual incidence in cirrhotic patients: 3-4% per year
- 20-year cumulative incidence: ~30%
- Risk persists despite normalization of iron stores (oxidative DNA damage irreversible)
- Surveillance (6-monthly ultrasound + AFP) enables early detection and curative intervention (resection, ablation, transplantation)
- HCC in non-cirrhotic haemochromatosis extremely rare [12,18]
Cardiomyopathy:
- If detected early (cardiac T2* 10-20 ms, asymptomatic or mild symptoms): Intensive iron depletion (weekly phlebotomy or chelation) can improve or normalize cardiac function over 6-24 months
- If severe (T2* less than 10 ms, symptomatic heart failure): Poor prognosis; heart failure may be irreversible; cardiac transplantation rarely required
- Juvenile haemochromatosis (type 2): Cardiomyopathy leading cause of death if untreated; aggressive treatment essential [19]
Diabetes Mellitus:
- Often irreversible despite iron depletion (β-cell destruction)
- Glycemic control may improve with phlebotomy in some patients
- Standard diabetes complications (retinopathy, nephropathy, neuropathy, cardiovascular disease) apply
- Requires lifelong management [2,10]
Arthropathy:
- Does not improve with iron depletion (cartilage damage and chondrocalcinosis irreversible)
- Progressive in ~20-30% despite treatment
- May require arthroplasty (hip, knee) for severe joint destruction
- Most disabling irreversible complication in well-treated patients [20]
Hypogonadism:
- Usually persists despite iron depletion (pituitary damage irreversible)
- Requires lifelong testosterone/estrogen replacement
- Fertility may remain impaired (specialist reproductive endocrinology input if conception desired) [2,10]
Long-Term Outcomes with Adherence
Maintenance Phlebotomy Adherence:
- Excellent adherence (ferritin maintained 50-100 μg/L): Normal life expectancy, minimal symptoms
- Poor adherence (iron reaccumulation): Progressive organ damage; cirrhosis risk if ferritin chronically > 1000 μg/L over years
- Most patients tolerate lifelong maintenance venesection well (every 2-4 months) [3,4]
Survival Data
Historical (Pre-Treatment Era):
- Median survival from symptom onset: less than 5 years
- Mortality from cirrhosis, HCC, heart failure, infections [2]
Contemporary (With Treatment):
- Non-cirrhotic at diagnosis: Survival equivalent to age-matched general population
- Cirrhotic at diagnosis: 5-year survival ~50-60%; 10-year survival ~30-40%
- Leading causes of death in treated cirrhotic patients: HCC (40-50%), hepatic decompensation (20-30%), cardiovascular disease (10-20%) [11,12]
Predictors of Survival
Favorable Prognostic Factors:
- Age less than 40 at diagnosis
- Absence of cirrhosis
- Ferritin less than 1000 μg/L at diagnosis
- No diabetes or cardiac involvement
- Female sex
- Adherence to maintenance phlebotomy [2,10,11]
Adverse Prognostic Factors:
- Cirrhosis at diagnosis (most important)
- Age > 50 at diagnosis
- Ferritin > 1000 μg/L
- Hepatomegaly, diabetes, cardiomyopathy present
- Alcohol consumption
- Concurrent liver disease (HCV, HBV, NAFLD) [2,10,11]
Quality of Life
- Pre-treatment symptoms (fatigue, arthralgia) often significantly impair quality of life
- Fatigue typically improves within 3-6 months of starting phlebotomy
- Skin pigmentation gradually fades over months-years
- Arthropathy remains the most common persistent symptom affecting quality of life in otherwise well-treated patients
- Sexual function may improve with testosterone replacement (males) if hypogonadism treated
- Overall, quality of life after treatment comparable to general population if organ damage prevented [2,6]
10. Examination Focus
Viva Points
Opening Statement:
"Hereditary haemochromatosis is an autosomal recessive disorder of iron metabolism, predominantly caused by homozygous C282Y mutation in the HFE gene. It results in inappropriately low hepcidin synthesis, leading to unregulated intestinal iron absorption and progressive parenchymal iron deposition in the liver, pancreas, heart, joints, and pituitary gland. It is the most common inherited metabolic disorder in Northern European populations, with C282Y homozygosity affecting approximately 1 in 200-300 individuals. The hallmark biochemical findings are elevated transferrin saturation greater than 45% and elevated ferritin. Treatment is with therapeutic phlebotomy to normalize iron stores, which prevents organ damage and ensures normal life expectancy if initiated before cirrhosis develops."
Key Facts to Mention:
-
Genetics: Autosomal recessive; C282Y homozygosity (85% of cases); chromosome 6 HFE gene; incomplete penetrance (1-10% of male homozygotes develop clinical disease)
-
Pathophysiology: HFE mutation → low hepcidin → increased ferroportin activity → enhanced intestinal iron absorption → progressive parenchymal iron deposition → oxidative damage
-
Epidemiology: 1:200-300 Northern Europeans are C282Y homozygotes; 1:8-10 carriers; male:female clinical expression 3-5:1; presents age 40-60 (men), 50-70 (women)
-
Clinical Features: Classic triad "bronze diabetes" (cirrhosis, diabetes, bronze skin) now rare; more commonly asymptomatic or fatigue/arthralgia; arthropathy (MCP 2/3 characteristic)
-
Diagnosis: Transferrin saturation > 45% (best screening test); elevated ferritin; HFE genotyping (C282Y/C282Y confirms); liver MRI R2* quantifies iron; liver biopsy rarely needed
-
Management: Therapeutic phlebotomy - induction (500 mL weekly until ferritin 50-100 μg/L), maintenance (every 2-4 months lifelong); target ferritin 50-100 μg/L
-
Complications: Cirrhosis (5-10% at diagnosis contemporary cohorts); HCC risk 20-30% if cirrhotic (persists despite iron depletion → lifelong 6-monthly surveillance); diabetes 30-60%; cardiomyopathy 15-30%; arthropathy 40-50% (irreversible)
-
Prognosis: Normal life expectancy if treated before cirrhosis; 5-year survival ~50-60% if cirrhotic at diagnosis; HCC leading cause of death in cirrhotic patients
-
Family Screening: Screen all first-degree relatives with HFE genotyping and iron studies (siblings 25% risk of homozygosity)
-
Evidence: EASL 2010 guidelines [PMID: 20471131]; AASLD 2011 guidelines [PMID: 21452290]; transferrin saturation > 45% has ~94% sensitivity for HH [3,4]
Common Exam Questions
Q1: "A 52-year-old man presents with fatigue and is found to have ferritin 1500 μg/L and transferrin saturation 75%. How would you investigate and manage this patient?"
Model Answer:
"This presentation suggests iron overload, most likely hereditary haemochromatosis given the markedly elevated transferrin saturation. My approach would be systematic:
Confirm Iron Overload:
- Repeat iron studies (transferrin saturation and ferritin) fasting to confirm persistence
- Exclude secondary causes: full blood count (anemia suggesting transfusion-related overload or chronic hemolysis), liver function tests, CRP (ferritin is an acute phase reactant), alcohol history, viral hepatitis serology
- Review medications (oral or intravenous iron supplements)
HFE Genotyping:
- Test for C282Y and H63D mutations
- C282Y homozygosity would confirm HFE haemochromatosis diagnosis (85% probability in this clinical scenario)
Assess Extent of Iron Loading:
- Liver MRI R2* or T2* to quantify hepatic iron concentration
- If uncertainty or very high ferritin (> 1000 μg/L), consider liver biopsy to assess both iron load and fibrosis stage
Assess Organ Damage:
- Liver: LFTs, abdominal ultrasound, transient elastography (FibroScan) to assess fibrosis; if cirrhosis suspected, endoscopy for varices and commence HCC surveillance
- Cardiac: ECG, echocardiogram; if age less than 50 or very high ferritin, cardiac MRI T2*
- Endocrine: Fasting glucose, HbA1c, testosterone, LH, FSH, thyroid function
- Joints: X-rays of hands (MCP joints) if symptomatic; DEXA scan for osteoporosis
Management:
- Initiate therapeutic phlebotomy: 500 mL weekly, checking hemoglobin before each session, ferritin every 3 months
- Target ferritin 50-100 μg/L (may take 6-24 months depending on total iron burden)
- Once target reached, switch to maintenance phlebotomy every 2-4 months lifelong
- Lifestyle advice: avoid iron and vitamin C supplements, minimize alcohol
- If cirrhosis confirmed: lifelong 6-monthly HCC surveillance (ultrasound + AFP)
Family Screening:
- Offer HFE genotyping to all first-degree relatives (siblings 25% risk if both parents carriers)
Prognosis:
- If no cirrhosis: normal life expectancy with adherence to phlebotomy
- If cirrhosis present: increased HCC risk necessitating surveillance; 5-year survival ~50-60%"
Q2: "Why is transferrin saturation the best screening test for haemochromatosis?"
Model Answer:
"Transferrin saturation is superior to ferritin for screening because it reflects circulating iron and becomes elevated early in the disease process, before tissue iron accumulation causes ferritin elevation.
Transferrin saturation represents the proportion of transferrin-binding sites occupied by iron. In haemochromatosis, inappropriately low hepcidin allows unregulated iron absorption, leading to progressive saturation of transferrin. Once transferrin saturation exceeds approximately 45%, non-transferrin-bound iron (NTBI) appears in plasma and is readily taken up by parenchymal tissues. Transferrin saturation > 45% has approximately 94% sensitivity and 86% specificity for HFE haemochromatosis.
In contrast, ferritin is an acute phase reactant and is elevated by inflammation, infection, malignancy, liver disease, and metabolic syndrome. While ferritin reflects body iron stores in the absence of confounding factors, it is less specific for haemochromatosis. Additionally, ferritin rises later in the disease course, after significant iron accumulation has occurred.
Therefore, the recommended screening approach is fasting transferrin saturation. If persistently > 45%, proceed to HFE genotyping. Ferritin is measured alongside to assess the degree of iron accumulation and guide treatment intensity."
Q3: "Explain the molecular basis of haemochromatosis and the role of hepcidin."
Model Answer:
"Hereditary haemochromatosis results from mutations in genes regulating the hepcidin-ferroportin axis, the master regulator of systemic iron homeostasis.
Normal Hepcidin-Ferroportin Regulation: Hepcidin is a 25-amino acid peptide hormone synthesized by hepatocytes in response to iron sufficiency. Hepcidin binds to ferroportin, the sole cellular iron exporter located on duodenal enterocytes, macrophages, and hepatocytes. Hepcidin binding triggers ferroportin internalization and degradation, thereby reducing iron export into plasma.
HFE Protein Function: Wild-type HFE protein complexes with β2-microglobulin and interacts with transferrin receptor 1 (TFR1) on hepatocyte membranes. When transferrin-bound iron increases, it displaces the HFE-TFR1 complex, allowing HFE to interact with transferrin receptor 2 (TFR2). This activates the BMP-SMAD signaling pathway (particularly BMP6), upregulating hepcidin transcription.
C282Y Mutation Effect: The C282Y mutation (cysteine to tyrosine at position 282) disrupts a critical disulfide bond, preventing proper HFE-β2-microglobulin complex formation. Mutant HFE is retained in the endoplasmic reticulum and degraded. This abolishes the iron-sensing mechanism, resulting in inappropriately low hepcidin synthesis despite iron overload.
Pathophysiological Consequence: Low hepcidin fails to downregulate ferroportin. This leads to:
- Enhanced duodenal iron absorption (2-3× normal)
- Continued macrophage iron recycling from senescent erythrocytes
- Progressive transferrin saturation
- Non-transferrin-bound iron formation when transferrin saturates
- Parenchymal cell iron uptake (liver, heart, pancreas, pituitary, joints)
- Iron-catalyzed oxidative damage (Fenton reaction generating hydroxyl radicals)
- Organ dysfunction: hepatic fibrosis, cardiomyopathy, diabetes, arthropathy
Non-HFE Forms: Mutations in HJV (hemojuvelin) or HAMP (hepcidin gene itself) directly impair hepcidin production, causing severe juvenile haemochromatosis. TFR2 mutations disrupt the iron-sensing pathway similarly to HFE. SLC40A1 (ferroportin gene) mutations cause a distinct autosomal dominant pattern with elevated ferritin but normal/low transferrin saturation."
Q4: "A patient with known cirrhosis from haemochromatosis has successfully completed iron depletion with ferritin now 60 μg/L. Does he still need HCC surveillance? Why?"
Model Answer:
"Yes, absolutely. This patient requires lifelong hepatocellular carcinoma surveillance with 6-monthly abdominal ultrasound and alpha-fetoprotein despite complete iron depletion.
Rationale: The HCC risk in haemochromatosis is predominantly a consequence of cirrhosis, not iron per se. Once cirrhosis is established, the architectural distortion, chronic inflammation, and mutagenic burden persist despite normalization of iron stores. Studies demonstrate that the annual incidence of HCC in cirrhotic haemochromatosis patients is 3-4% per year, with a 20-year cumulative incidence of approximately 30%. This represents a 200-fold increased risk compared to the general population.
Critically, this elevated HCC risk persists even after complete iron depletion. The oxidative DNA damage caused by iron-catalyzed Fenton reactions during the period of iron overload is irreversible, with accumulated mutations predisposing to malignant transformation. Additionally, cirrhosis itself creates a pro-carcinogenic environment through chronic inflammation, hepatocyte regeneration, and dysplasia.
Evidence: Landmark studies (e.g., Niederau et al.) demonstrated that cirrhotic haemochromatosis patients who achieved complete iron depletion still had a 30% cumulative HCC incidence over 10 years, compared to less than 1% in non-cirrhotic treated patients. This evidence base underpins guidelines recommending lifelong HCC surveillance in all cirrhotic patients regardless of iron status.
Surveillance Protocol:
- Abdominal ultrasound every 6 months
- Serum AFP every 6 months (although sensitivity limited, combined with ultrasound improves detection)
- Continue indefinitely, lifelong
- If nodule detected: triphasic CT or MRI liver for characterization; multidisciplinary hepatobiliary team discussion
Contrast with Non-Cirrhotic Patients: Patients treated before cirrhosis develops have HCC risk equivalent to the general population and do not require surveillance.
Therefore, the key determinant of HCC risk is the presence of cirrhosis, not current iron status. This patient must continue surveillance indefinitely."
Q5: "What is the inheritance pattern of haemochromatosis and how would you counsel a C282Y homozygote patient about family screening?"
Model Answer:
"Hereditary haemochromatosis caused by HFE mutations follows an autosomal recessive inheritance pattern.
Genetic Counseling:
Patient's Relatives: I would explain that as a C282Y homozygote, the patient has inherited one C282Y mutation from each parent. This means both parents are obligate carriers (C282Y heterozygotes). Siblings have:
- 25% risk of being C282Y homozygotes (affected)
- 50% risk of being heterozygous carriers (C282Y/wild-type)
- 25% risk of being unaffected (wild-type/wild-type)
Recommendation: All first-degree relatives (parents, siblings, children) should undergo HFE genotyping and iron studies (transferrin saturation and ferritin). This allows early identification of at-risk homozygous relatives before organ damage occurs.
Patient's Children: All children of a C282Y homozygote will be at least carriers (C282Y heterozygotes), having inherited one C282Y allele from the affected parent. Whether they are homozygotes depends on the partner's genotype:
- If partner is wild-type (most likely): all children are carriers (C282Y heterozygotes) - not at risk of disease
- If partner is a carrier (1:8-10 in Northern Europeans): 50% of children are homozygotes (C282Y/C282Y) - at risk of disease; 50% are carriers
- If partner is a homozygote (rare): all children are homozygotes
Partner Screening: I would offer HFE genotyping to the patient's partner, particularly if planning children or if of Northern European ancestry (higher carrier frequency). If the partner is a carrier, children have 50% risk of homozygosity and should be monitored.
Incomplete Penetrance: I would emphasize that C282Y homozygosity does not guarantee clinical disease. Only approximately 1-10% of male homozygotes and 0.5-2% of female homozygotes develop clinically significant iron overload requiring treatment. Biochemical penetrance (elevated iron studies) is higher (40-70% males, 10-30% females), but many remain asymptomatic. Therefore, genotype-positive relatives require periodic iron studies (initially, then every 2-5 years) even if normal at baseline.
Treatment: I would reassure that if relatives are found to be homozygotes with iron overload, treatment with phlebotomy is highly effective, and if started before cirrhosis develops, ensures normal life expectancy.
Practical Approach:
- Recommend all first-degree relatives contact their GP for HFE genotyping and iron studies
- Provide written information about haemochromatosis and inheritance
- If siblings/children are C282Y homozygotes, refer to hepatology for assessment and management
- Emphasize the importance of early detection to prevent complications"
Q6: "How does iron cause organ damage in haemochromatosis?"
Model Answer:
"Iron causes organ damage in haemochromatosis primarily through oxidative stress mediated by the Fenton reaction, with subsequent cellular and molecular damage.
Oxidative Stress Mechanism: Free intracellular iron (Fe²⁺) catalyzes the Fenton reaction: Fe²⁺ + H₂O₂ → Fe³⁺ + OH· + OH⁻. This generates highly reactive hydroxyl radicals (OH·), which cause:
- Lipid peroxidation: Attack on polyunsaturated fatty acids in cell membranes, disrupting membrane integrity and causing cell lysis
- DNA damage: Hydroxyl radicals cause DNA strand breaks, base modifications, and mutations; mutagenic effect predisposes to hepatocellular carcinoma
- Protein oxidation: Carbonylation and cross-linking of proteins, impairing enzymatic function
- Mitochondrial dysfunction: Damage to mitochondrial membranes and respiratory chain enzymes, reducing ATP production and increasing apoptosis
Organ-Specific Mechanisms:
Liver:
- Hepatocyte iron accumulation → oxidative stress → stellate cell activation
- Stellate cells transform into collagen-producing myofibroblasts
- Progressive extracellular matrix deposition → bridging fibrosis → cirrhosis
- Chronic inflammation + oxidative DNA damage → dysplasia → hepatocellular carcinoma
Pancreas:
- Iron deposition in pancreatic β-cells → oxidative damage → apoptosis
- Progressive loss of insulin-secreting capacity → diabetes mellitus (typically insulin-requiring)
- Exocrine pancreas relatively spared (clinically significant pancreatic insufficiency rare)
Heart:
- Myocardial iron accumulation → cardiomyocyte dysfunction
- Initially diastolic dysfunction (restrictive pattern)
- Progressive systolic dysfunction (dilated cardiomyopathy)
- Conduction system damage → arrhythmias (atrial fibrillation, heart block, ventricular arrhythmias)
- Severe iron loading → intractable heart failure
Pituitary:
- Iron deposition in gonadotrophs → hypogonadotropic hypogonadism (low LH, FSH)
- Results in low testosterone (males) or amenorrhea (females)
- Loss of libido, erectile dysfunction, infertility
- Other pituitary hormones usually spared (GH, ACTH, TSH)
Joints:
- Iron deposition in synovium → calcium pyrophosphate crystal formation
- Chondrocalcinosis (visible on X-ray)
- Cartilage degradation → osteoarthritis-like picture
- Particularly affects metacarpophalangeal joints (2nd/3rd MCP characteristic)
- Acute pseudogout flares (calcium pyrophosphate arthropathy)
Skin:
- Iron deposition in dermis combined with increased melanin synthesis
- Bronze or slate-grey hyperpigmentation
Pattern of Iron Deposition: Unlike secondary iron overload (transfusional), HFE haemochromatosis causes predominantly parenchymal (hepatocyte, pancreatic β-cell, cardiomyocyte) iron loading, with relative sparing of reticuloendothelial macrophages (Kupffer cells, splenic macrophages). This reflects the mechanism: increased absorption and ferroportin-mediated iron export from macrophages into circulation → parenchymal uptake.
Irreversibility:
- DNA mutations are irreversible (explains persistent HCC risk after iron depletion)
- Cirrhosis architectural distortion irreversible (fibrosis may partially regress if non-cirrhotic)
- Cartilage damage irreversible (arthropathy does not improve with iron depletion)
- β-cell loss and pituitary damage often irreversible (diabetes and hypogonadism persist)
Preventability: Early iron depletion before accumulation reaches toxic threshold prevents oxidative damage and organ dysfunction, underscoring the importance of early diagnosis."
Common Mistakes to Avoid
❌ Mistake 1: Dismissing mildly elevated ferritin (200-500 μg/L) without checking transferrin saturation
- Ferritin can be mildly elevated in metabolic syndrome, inflammation, or liver disease
- Always measure transferrin saturation (best screening test)
- If TfSat > 45%, proceed to HFE genotyping regardless of ferritin level
❌ Mistake 2: Diagnosing haemochromatosis based on C282Y heterozygosity
- C282Y heterozygotes (C282Y/wild-type) are carriers, not affected
- Carrier frequency is 1:8-10 in Northern Europeans
- Heterozygotes do not develop HFE haemochromatosis (autosomal recessive)
- If iron overload present in heterozygote, investigate other causes (secondary overload, non-HFE mutations, metabolic syndrome)
❌ Mistake 3: Stopping HCC surveillance in cirrhotic patients after iron depletion
- HCC risk persists despite normalization of iron stores
- Annual incidence remains 3-4% per year in cirrhotic patients
- Lifelong 6-monthly ultrasound + AFP mandatory
- Non-cirrhotic patients do not require HCC surveillance
❌ Mistake 4: Expecting arthropathy to improve with phlebotomy
- Arthropathy is the only major complication that does not improve with iron depletion
- Established chondrocalcinosis and cartilage damage are irreversible
- Inform patients to manage expectations
- Phlebotomy may prevent progression but does not reverse established joint disease
❌ Mistake 5: Failing to screen first-degree relatives
- Siblings have 25% risk of being C282Y homozygotes
- Early detection in asymptomatic relatives prevents organ damage
- Recommend HFE genotyping and iron studies for all first-degree relatives
- Optimal screening before age 30-40 years
❌ Mistake 6: Attributing all elevated ferritin to haemochromatosis
- Ferritin is an acute phase reactant (infection, inflammation, malignancy)
- Hyperferritinemia without iron overload is common (metabolic syndrome, chronic liver disease)
- Check transferrin saturation: normal or low TfSat suggests non-iron cause
- CRP, liver function tests, clinical context essential
❌ Mistake 7: Over-restricting dietary iron in diagnosed patients
- Phlebotomy removes far more iron than dietary restriction can achieve
- Moderate dietary iron intake acceptable (phlebotomy compensates)
- Avoid extreme restriction (nutritionally unnecessary and poor adherence)
- Do avoid iron supplements and excess vitamin C; minimize alcohol
❌ Mistake 8: Using liver biopsy routinely for diagnosis
- HFE genotyping confirms diagnosis in > 95% of cases (Northern Europeans)
- Liver MRI R2* quantifies hepatic iron non-invasively
- Biopsy reserved for: uncertainty about diagnosis, very high ferritin (> 1000 μg/L) needing fibrosis staging, suspected concurrent liver disease
- Non-invasive approaches now standard
11. References
-
European Association for the Study of the Liver. EASL Clinical Practice Guidelines for HFE Hemochromatosis. J Hepatol. 2010;53(1):3-22. doi:10.1016/j.jhep.2010.03.001
-
Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diagnosis and Management of Hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(1):328-343. doi:10.1002/hep.24330
-
Adams PC, Barton JC. A diagnostic approach to hyperferritinemia with a non-elevated transferrin saturation. J Hepatol. 2011;55(2):453-458. doi:10.1016/j.jhep.2011.02.010
-
Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139(2):393-408. doi:10.1053/j.gastro.2010.06.013
-
Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016;388(10045):706-716. doi:10.1016/S0140-6736(15)01315-X
-
Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358(3):221-230. doi:10.1056/NEJMoa073286
-
Hanson EH, Imperatore G, Burke W. HFE gene and hereditary hemochromatosis: a HuGE review. Am J Epidemiol. 2001;154(3):193-206. doi:10.1093/aje/154.3.193
-
Waalen J, Felitti VJ, Gelbart T, Beutler E. Screening for hemochromatosis by measuring ferritin levels: a more effective approach. Blood. 2008;111(7):3373-3376. doi:10.1182/blood-2007-07-102673
-
Pankow JS, Boerwinkle E, Adams PC, et al. HFE C282Y homozygotes have reduced low-density lipoprotein cholesterol: the Atherosclerosis Risk in Communities (ARIC) Study. Transl Res. 2008;152(1):3-10. doi:10.1016/j.trsl.2008.05.005
-
Niederau C, Fischer R, Pürschel A, Stremmel W, Häussinger D, Strohmeyer G. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology. 1996;110(4):1107-1119. doi:10.1053/gast.1996.v110.pm8613000
-
Falize L, Guillygomarc'h A, Perrin M, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology. 2006;44(2):472-477. doi:10.1002/hep.21260
-
Elmberg M, Hultcrantz R, Ekbom A, et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology. 2003;125(6):1733-1741. doi:10.1053/j.gastro.2003.09.035
-
Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399-408. doi:10.1038/ng0896-399
-
Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717. doi:10.1182/blood-2005-05-1857
-
Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823(9):1434-1443. doi:10.1016/j.bbamcr.2012.01.014
-
Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7(3):205-214. doi:10.1016/j.cmet.2007.11.016
-
Britton RS, Leicester KL, Bacon BR. Iron toxicity and chelation therapy. Int J Hematol. 2002;76(3):219-228. doi:10.1007/BF02982791
-
Kowdley KV, Belt P, Wilson LA, et al. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2012;55(1):77-85. doi:10.1002/hep.24706
-
Murphy CJ, Oudit GY. Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment. J Card Fail. 2010;16(11):888-900. doi:10.1016/j.cardfail.2010.05.009
-
Carroll GJ, Breidahl WH, Bulsara MK, Olynyk JK. Hereditary hemochromatosis is characterized by a clinically definable arthropathy that correlates with iron load. Arthritis Rheum. 2011;63(1):286-294. doi:10.1002/art.30094
-
Wood JC, Enriquez C, Ghugre N, et al. MRI R2 and R2* mapping accurately estimates hepatic iron concentration in transfusion-dependent thalassemia and sickle cell disease patients. Blood. 2005;106(4):1460-1465. doi:10.1182/blood-2004-10-3982
-
Bullen JJ, Rogers HJ, Spalding PB, Ward CG. Iron and infection: the heart of the matter. FEMS Immunol Med Microbiol. 2005;43(3):325-330. doi:10.1016/j.femsim.2004.11.010
Last Updated: 2026-01-05
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Iron Metabolism
Differentials
Competing diagnoses and look-alikes to compare.
- Secondary Iron Overload
- Porphyria Cutanea Tarda
- Non-Transferrin Bound Iron Disorders
Consequences
Complications and downstream problems to keep in mind.
- Cirrhosis
- Hepatocellular Carcinoma
- Dilated Cardiomyopathy