IgG4-Related Disease
IgG4-RD typically affects middle-aged to elderly men with a median age of 60 years and a male-to-female ratio of 2-3:1, though head and neck involvement shows equal sex distribution. The disease is characterised by...
What matters first
IgG4-RD typically affects middle-aged to elderly men with a median age of 60 years and a male-to-female ratio of 2-3:1, though head and neck involvement shows equal sex distribution. The disease is characterised by...
Biliary obstruction requiring urgent decompression
9 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Biliary obstruction requiring urgent decompression
- Aortic aneurysm with wall thickening (risk of rupture)
- Orbital mass with proptosis causing vision loss
- Acute renal failure from tubulointerstitial nephritis
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
IgG4-Related Disease
1. Clinical Overview
Summary
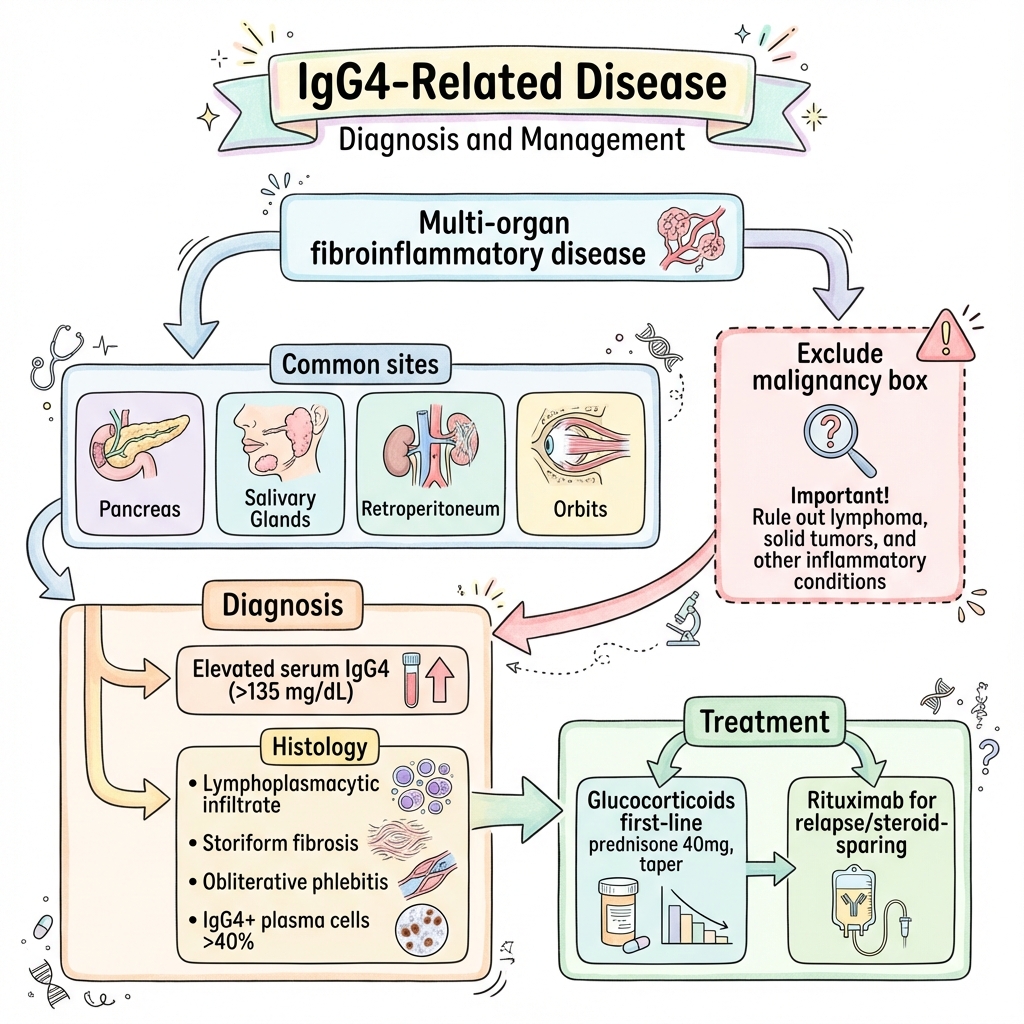
IgG4-related disease (IgG4-RD) is a fibroinflammatory condition characterised by tumefactive lesions, dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform (cartwheel-pattern) fibrosis, and obliterative phlebitis. First recognised as a unified disease entity in 2003, IgG4-RD encompasses conditions previously described in individual organs including autoimmune pancreatitis type 1, Mikulicz disease, Riedel thyroiditis, and retroperitoneal fibrosis. The disease can affect virtually any organ system, most commonly the pancreas, biliary tree, salivary glands, lacrimal glands, retroperitoneum, kidneys, aorta, and meninges. IgG4-RD is increasingly recognized as a cause of significant morbidity and mortality worldwide, with advances in pathophysiology understanding leading to improved diagnostic and therapeutic approaches. [1,2,23]
IgG4-RD typically affects middle-aged to elderly men with a median age of 60 years and a male-to-female ratio of 2-3:1, though head and neck involvement shows equal sex distribution. The disease is characterised by two distinct clinical phenotypes: proliferative (inflammatory) manifestations with organ enlargement and active inflammation, and fibrotic manifestations with established scarring and organ dysfunction. These phenotypes frequently coexist, creating diagnostic challenges. [3,4]
The pathogenesis involves aberrant adaptive immune responses with expansion of CD4+ cytotoxic T lymphocytes (CD4 CTLs), T follicular helper 2 cells (Tfh2), and IgG4-switched plasmablasts. Multiple autoantigens have been identified including galectin-3, laminin 511-E8, prohibitin, and annexin A11, supporting an autoimmune pathogenesis. The remarkable responsiveness to B cell depletion with rituximab confirms the central role of B cells in disease pathology. Recent studies have identified distinct immunological phenotypes based on clinical presentation, with retroperitoneal/aortitis phenotypes dominated by CX3CR1+ cytotoxic CD4+ T cells, while Mikulicz disease with systemic involvement shows predominance of Tfh2 cells and higher relapse risk. [5,6,24]
Diagnosis requires integration of clinical presentation, serological findings (elevated serum IgG4 > 135 mg/dL in 60-70%), characteristic radiological features, and histopathological confirmation showing the triad of dense lymphoplasmacytic infiltrate, storiform fibrosis, and obliterative phlebitis with IgG4+ plasma cells comprising > 40% of IgG+ cells. The 2019 ACR/EULAR classification criteria provide a validated framework for diagnosis with specificity 99.2% and sensitivity 85.5%. [7,8]
First-line treatment consists of glucocorticoid therapy (prednisone 0.6 mg/kg/day, typically 40 mg daily, with taper over 3-6 months), which produces dramatic responses in > 90% of patients, often within days to weeks. However, relapse rates of 30-60% are common, particularly after steroid withdrawal. Rituximab has emerged as highly effective second-line therapy for relapsing disease, steroid-refractory cases, and maintenance of remission, with the MITIGATE trial providing definitive evidence of B cell depletion efficacy. In 2025, rituximab received regulatory approval for IgG4-RD in select jurisdictions, marking a major milestone in disease management. Early treatment is critical to prevent irreversible fibrosis and organ dysfunction. [9,10,11,25,26]
The major diagnostic challenge is differentiation from malignancy, particularly pancreatic cancer, cholangiocarcinoma, and retroperitoneal sarcoma. IgG4-RD can closely mimic these malignancies both clinically and radiologically, making tissue diagnosis essential before initiating immunosuppression. Other important differentials include primary sclerosing cholangitis, sarcoidosis, Sjögren syndrome, and systemic vasculitis. [12,13]
Key Facts
- Definition: Immune-mediated fibroinflammatory condition characterised by IgG4+ plasma cell infiltration, storiform fibrosis, and obliterative phlebitis
- Incidence: Estimated 0.8-1.0 per 100,000 per year; increasingly recognised, likely underdiagnosed
- Peak Demographics: Median age 60 years (range 40-80); male predominance 2-3:1 (except head/neck involvement)
- Common Organs: Pancreas (autoimmune pancreatitis type 1), biliary tree, salivary glands, lacrimal glands, retroperitoneum, kidneys, aorta, lymph nodes, meninges
- Pathognomonic Features: Storiform fibrosis + obliterative phlebitis + IgG4+ plasma cells > 40% of IgG+ cells (> 10-50 per high-power field depending on organ)
- Serum IgG4: Elevated (> 135 mg/dL or > 1.35 g/L) in only 60-70%; normal levels do not exclude diagnosis
- Gold Standard Investigation: Histopathology with IgG4 and IgG immunostaining from affected organ biopsy
- Classification Criteria: 2019 ACR/EULAR criteria with 99.2% specificity, 85.5% sensitivity using weighted scoring system
- First-line Treatment: Glucocorticoids (prednisone 40 mg/day or 0.6 mg/kg/day, taper over 3-6 months)
- Second-line Treatment: Rituximab (1g IV repeated after 2 weeks, or 375 mg/m² weekly × 4)
- Steroid Response: Rapid and dramatic improvement in > 90%; lack of response should prompt diagnostic reconsideration
- Relapse Rate: 30-60% after glucocorticoid withdrawal; higher with single-organ involvement and rapid steroid taper
- Prognosis: Excellent with treatment; mortality primarily from untreated complications (organ failure, aneurysm rupture)
Clinical Pearls
Diagnostic Pearl: Serum IgG4 elevation alone is insufficient for diagnosis—it is elevated in only 60-70% of IgG4-RD patients and can be elevated in malignancy, infection, and other inflammatory conditions. Histopathological confirmation is essential. [14]
Treatment Pearl: Response to glucocorticoids is typically rapid (days to weeks) and dramatic. Absence of clinical or radiological improvement within 2-4 weeks should prompt reconsideration of the diagnosis and exclusion of malignancy. [9]
Pitfall Warning: IgG4-RD is the great mimicker—pancreatic, biliary, and retroperitoneal manifestations closely resemble malignancy. Always obtain tissue diagnosis before initiating immunosuppression to avoid missing cancer. [12,13]
Phenotype Recognition: IgG4-RD presents with two overlapping phenotypes: proliferative (soft, inflammatory organ enlargement responsive to steroids) and fibrotic (established scarring with limited reversibility). Early treatment of proliferative disease prevents progression to irreversible fibrosis. [3]
Rituximab Advantage: For relapsing disease, rituximab maintenance every 6 months achieves superior remission rates (> 80%) compared to conventional immunosuppressants and avoids long-term steroid toxicity. [15,16]
Multi-organ Surveillance: At diagnosis, screen for involvement of other organs even if asymptomatic—60% have multi-organ involvement. Use CT chest/abdomen/pelvis and organ-specific investigations based on clinical suspicion. [4]
Mnemonic: STORIFORM - Storiform fibrosis, Tissue infiltration (lymphoplasmacytic), Obliterative phlebitis, Responsive to steroids, IgG4 plasma cells, Fibrosis can be irreversible, Organs multiple, Relapse common, Mimics malignancy
Why This Matters Clinically
IgG4-RD represents a paradigm shift in our understanding of fibroinflammatory disease. Its recognition prevents unnecessary surgeries for presumed malignancy and enables effective immunosuppressive treatment. Unlike many fibrotic disorders (systemic sclerosis, idiopathic pulmonary fibrosis), IgG4-RD fibrosis can be partially reversible with early treatment, making prompt diagnosis critical.
The disease challenges traditional organ-based medicine—patients often see multiple specialists (gastroenterologists for pancreatitis, urologists for retroperitoneal fibrosis, ophthalmologists for orbital disease) before the systemic nature is recognised. Awareness of IgG4-RD enables earlier diagnosis and prevents progression to irreversible organ damage.
For postgraduate examinations (MRCP, FRACP), IgG4-RD is a high-yield topic appearing in data interpretation stations (elevated IgG4, imaging showing "sausage-shaped" pancreas), clinical cases (painless jaundice, salivary gland swelling), and viva voce scenarios (differential diagnosis of pancreatic masses, approach to retroperitoneal fibrosis).
2. Epidemiology
Incidence and Prevalence
The reported incidence of IgG4-RD is approximately 0.8-1.0 per 100,000 per year based on population studies from Japan and Europe. For autoimmune pancreatitis (the most studied manifestation), incidence is 0.8 per 100,000. True incidence is likely higher due to underrecognition and evolving diagnostic criteria. IgG4-RD affects patients of diverse racial and ethnic backgrounds globally, though higher recognition rates in Asian populations may reflect referral and diagnostic bias rather than true ethnic predisposition. [17,27]
Prevalence estimates range from 3.5 to 6.0 per 100,000 population. IgG4-RD is increasingly recognised worldwide, with substantial case series reported from Japan, North America, Europe, and increasingly from other regions as awareness grows. [18]
Demographics
| Factor | Details |
|---|---|
| Age | Median 60 years; range 40-80 years; rare in children |
| Sex | Overall M:F ratio 2-3:1; pancreatic/retroperitoneal 3-4:1; head/neck 1:1 |
| Ethnicity | Higher recognition in Japan; reported globally across all ethnic groups |
| Geographic | Described worldwide; possible Asian predominance may reflect recognition bias |
Risk Factors
Unlike many autoimmune diseases, IgG4-RD has few established risk factors:
Genetic Susceptibility:
- HLA associations reported: HLA-DRB10405 and HLA-DQB10401 in Japanese populations
- Familial clustering extremely rare
- Specific genetic markers have not been validated across populations [19]
Environmental Factors:
- No clear environmental triggers identified
- No consistent associations with infections, medications, or exposures
Associated Conditions:
- History of atopy reported in 30-40% (elevated IgE, peripheral eosinophilia)
- Possible increased risk in patients with other autoimmune conditions, though not clearly established
- Association with malignancy (lymphoma, carcinoma) reported but causality unclear [20]
Clinical Phenotypes
IgG4-RD can be classified into clinical phenotypes based on organ distribution patterns: [21]
- Pancreato-hepato-biliary phenotype: Type 1 autoimmune pancreatitis ± IgG4 sclerosing cholangitis
- Retroperitoneal fibrosis and/or aortitis phenotype: Dominated by CX3CR1+ cytotoxic CD4+ T cells
- Head and neck limited phenotype: Mikulicz disease (salivary/lacrimal), dacryoadenitis, sialadenitis
- Mikulicz disease with systemic involvement: Dominated by Tfh2 cells, higher relapse risk
These phenotypes have distinct immunological profiles and may guide treatment decisions. [6]
3. Pathophysiology
Molecular and Cellular Mechanisms
IgG4-RD pathogenesis involves complex interactions between innate and adaptive immunity, with B cells and specific CD4+ T cell subsets playing central roles. [5,6]
Step 1: Initiation and Antigenic Triggers
The initial trigger for IgG4-RD remains unknown. Current evidence suggests:
- Autoimmune mechanisms: Multiple autoantigens identified including galectin-3, laminin 511-E8, prohibitin, and annexin A11; in IgG4 sclerosing cholangitis, autoantibodies against annexin A11 and laminin 511-E8 have been specifically implicated in pathogenesis
- Molecular mimicry: Possible cross-reactivity between environmental antigens and self-antigens
- Loss of tolerance: Breakdown of immune regulation allowing self-reactive B and T cell expansion
- Genetic predisposition: HLA-DRB1 and HLA-DQB1 associations suggest MHC class II presentation of specific peptides; genome-wide studies continue to identify susceptibility loci [19,28]
Step 2: Adaptive Immune Activation
T Cell Responses:
- CD4+ Cytotoxic T Lymphocytes (CD4 CTLs): Express cytotoxic molecules (granzyme, perforin, CD25); infiltrate affected tissues and induce targeted apoptosis of host cells; particularly dominant in retroperitoneal/aortic phenotype [6]
- T Follicular Helper 2 Cells (Tfh2): Produce IL-4, IL-5, IL-13; drive IgG4 class-switching in B cells; dominant in Mikulicz disease with systemic involvement
- T Follicular Helper 1 Cells (Tfh1): Produce IFN-γ; induce IgG1 class-switching; associated with complement activation and hypocomplementemia in some patients
- Regulatory T Cells (Tregs): Expanded in IgG4-RD; produce IL-10 and TGF-β; paradoxically may promote fibrosis rather than suppress inflammation [5]
B Cell and Plasma Cell Responses:
- Oligoclonal expansion: IgG4+ plasmablasts show clonal expansion, suggesting antigen-driven selection
- IgG4 class-switching: Driven by Tfh2-derived IL-4 and IL-13 via CD40-CD40L interaction
- Autoantibody production: IgG4 and IgG1 autoantibodies against tissue-specific antigens
- Pathogenic role: Blocking autoantibodies (e.g., anti-laminin 511-E8) may impair epithelial protection against bile acids in cholangitis [22]
IgG4 Antibodies:
- IgG4 is generally considered non-pathogenic (cannot fix complement via classical pathway, weak Fc receptor binding, can undergo Fab-arm exchange)
- However, IgG4 autoantibodies may exert pathogenic effects through:
- Blocking protective mechanisms (anti-laminin 511-E8 in biliary disease impairs epithelial protection against bile acids)
- Immune complex formation (can activate complement via alternative pathway in high concentrations)
- Facilitation of tissue injury through other mechanisms
- Recent evidence suggests IgG4 antibodies in IgG4-RD may not always be bispecific due to Fab-arm exchange limitations, potentially enhancing pathogenicity [22,29]
Step 3: Tissue Infiltration
- Dense lymphoplasmacytic infiltrate accumulates in affected organs
- IgG4+ plasma cells typically comprise > 40% of total IgG+ plasma cells (> 10-50 per high-power field depending on organ)
- Eosinophil infiltration in 30-40% of cases
- Macrophage activation with M2 polarisation (pro-fibrotic phenotype)
Step 4: Fibrosis and Tissue Remodelling
Profibrotic Mechanisms:
- Cytokine-driven fibrosis: TGF-β, IL-4, IL-13 secreted by Th2 cells, Tregs, and macrophages activate fibroblasts
- Storiform fibrosis: Characteristic "cartwheel" or "matted" pattern of collagen deposition radiating from central nidus
- Obliterative phlebitis: Inflammatory infiltration and fibrotic occlusion of veins (pathognomonic feature)
- Exuberant wound healing: CD4 CTLs induce apoptosis → excessive tissue repair response → fibrosis [5]
Step 5: Organ Dysfunction
Mechanisms of Organ Damage:
- Mass effect: Tumefactive lesions causing compression (biliary obstruction, ureteric obstruction, orbital proptosis)
- Glandular destruction: Infiltration and fibrosis of salivary/lacrimal glands → sicca symptoms
- Vascular involvement: Aortitis with aneurysm formation, periarteritis
- Parenchymal replacement: Progressive fibrosis replacing functional tissue (pancreatic insufficiency, renal failure)
- Irreversible fibrosis: If untreated, inflammatory phase transitions to established fibrosis with limited reversibility
Histological Features (Consensus Criteria)
The 2012 Boston Consensus established histopathological criteria for IgG4-RD diagnosis: [23]
Major Criteria (Required for definitive diagnosis):
- Dense lymphoplasmacytic infiltrate: Sheets of lymphocytes and plasma cells
- Storiform fibrosis: Cartwheel or matted pattern of collagen fibers radiating from center
- Obliterative phlebitis: Vein lumen narrowing/occlusion by inflammatory infiltrate and fibrosis
Minor Criteria:
- IgG4+ plasma cells: > 10 per high-power field (HPF) in most organs; > 50/HPF for lymph nodes
- IgG4+/IgG+ ratio: > 40% in most organs; > 70% in lymph nodes
- Eosinophil infiltration: Present in 30-40% of cases
Organ-Specific Variations:
- Pancreas: Dense periductal lymphoplasmacytic infiltrate, obliterative phlebitis most prominent
- Biliary tree: Transmural inflammation, storiform fibrosis, IgG4+ cells around bile ducts
- Salivary glands: Lobular architecture preserved, periductal fibrosis, lymphoepithelial lesions rare (vs Sjögren)
- Kidney: Tubulointerstitial nephritis, tubular basement membrane thickening, glomeruli usually spared
4. Clinical Presentation
Organ-Specific Manifestations
IgG4-RD can affect virtually any organ. The most common presentations include: [4,24]
| Organ System | Specific Manifestation | Clinical Features | Key Imaging/Laboratory Findings |
|---|---|---|---|
| Pancreas | Type 1 autoimmune pancreatitis (AIP) | Painless obstructive jaundice (60%), epigastric discomfort, weight loss, new-onset diabetes (30%), steatorrhea if exocrine insufficiency | "Sausage-shaped" pancreas on CT/MRI, diffuse enlargement, delayed enhancement, capsule-like rim, pancreatic duct narrowing |
| Biliary Tree | IgG4 sclerosing cholangitis (IgG4-SC) | Jaundice, pruritus, cholangitis; mimics PSC and cholangiocarcinoma; found in 30-60% of type 1 AIP cases | Biliary strictures (intrahepatic > extrahepatic), wall thickening, elevated ALP/GGT, CA19-9 may be elevated |
| Salivary Glands | IgG4 sialadenitis (Küttner tumor, Mikulicz disease) | Bilateral painless parotid/submandibular swelling (80% bilateral), xerostomia, difficulty chewing/swallowing | Symmetric gland enlargement, homogeneous enhancement, reduced salivary flow |
| Lacrimal Glands | IgG4 dacryoadenitis | Bilateral painless lacrimal gland swelling, dry eyes, epiphora | Symmetric enlargement, homogeneous enhancement |
| Orbit | IgG4 orbital disease | Proptosis, diplopia (extraocular muscle involvement), periorbital swelling, vision loss (optic nerve compression) | Orbital soft tissue masses, extraocular muscle enlargement, lacrimal gland involvement |
| Retroperitoneum | Retroperitoneal fibrosis (RPF) | Back/flank pain, lower limb edema (IVC compression), symptoms of ureteric obstruction | Periaortic soft tissue mass ("mantle" around aorta), ureteric obstruction, hydronephrosis |
| Kidneys | IgG4 tubulointerstitial nephritis (IgG4-TIN), membranous nephropathy | Acute/subacute renal impairment, often asymptomatic, proteinuria, rarely nephrotic syndrome | Elevated creatinine, bilateral cortical lesions on CT (low-attenuation), IgG4-TIN on biopsy |
| Aorta/Large Vessels | IgG4 aortitis, periaortitis, inflammatory aneurysm | Often asymptomatic; may present with aneurysm, dissection, vascular claudication | Aortic wall thickening, periaortic soft tissue, aneurysm formation, FDG-PET avid |
| Lungs | IgG4 lung disease | Cough, dyspnea, or asymptomatic; nodular, mass-like, interstitial, or bronchovascular patterns | Pulmonary nodules, ground-glass opacities, interstitial infiltrates, mediastinal lymphadenopathy |
| Thyroid | Riedel thyroiditis | Woody hard thyroid, compressive symptoms (dysphagia, dyspnea, stridor), hypothyroidism | Thyroid fibrosis, often asymmetric, may extend to surrounding structures |
| Meninges | IgG4 pachymeningitis | Headache, cranial nerve palsies, seizures, focal neurological deficits | Dural thickening on MRI (often contrast-enhancing), mass-like lesions |
| Lymph Nodes | IgG4 lymphadenopathy | Painless lymphadenopathy, often mediastinal, hilar, or cervical | Enlarged lymph nodes, FDG-PET avid; biopsy shows IgG4+ plasma cells > 100/HPF |
| Pericardium | IgG4 pericarditis | Chest pain, pericardial effusion, constrictive physiology | Pericardial thickening/effusion on echo/CT |
| Skin | IgG4 cutaneous disease | Nodules, plaques, erythematous lesions | Dermal infiltration on biopsy |
| Breast | IgG4 mastitis | Breast mass, mimics carcinoma | Mass lesion on mammography/ultrasound |
Presentation Patterns
Common Initial Presentations:
- Painless obstructive jaundice: Most common in type 1 AIP with biliary involvement
- Bilateral gland swelling: Salivary/lacrimal gland enlargement (Mikulicz disease)
- Incidental mass: Found on imaging performed for other indications
- Renal impairment: Asymptomatic rise in creatinine from IgG4-TIN
- Sicca symptoms: Dry eyes/mouth from glandular involvement
- Subacute onset: Symptoms develop over weeks to months, not acute
Multi-Organ Involvement:
- Present in 60-90% of patients at diagnosis
- Sequential organ involvement over time in many patients
- Synchronous vs metachronous involvement (months to years apart)
- Higher relapse risk with multi-organ disease [4]
Constitutional Symptoms
Unlike many systemic inflammatory diseases, IgG4-RD typically presents with:
- Minimal systemic symptoms: Patients often appear well despite significant organ involvement
- Mild weight loss: Common with pancreatic involvement
- Absence of fever: Fever uncommon unless infection (e.g., cholangitis from biliary obstruction)
- Normal or mildly elevated inflammatory markers: CRP/ESR usually normal or slightly elevated (not highly inflammatory)
Red Flags
[!CAUTION] Critical Presentations Requiring Urgent Assessment
- Biliary obstruction: Ascending cholangitis, hepatic dysfunction, requires urgent ERCP ± stenting
- Aortic aneurysm: Risk of rupture, requires vascular surgery evaluation
- Acute renal failure: Rapidly progressive IgG4-TIN, may require dialysis
- Orbital mass with vision loss: Optic nerve compression, requires urgent treatment
- Hydronephrosis with declining renal function: Ureteric obstruction, may require stenting
- Mass suspicious for malignancy: Urgent biopsy to exclude cancer before immunosuppression
- Neurological deficits: IgG4 pachymeningitis or hypophysitis causing focal deficits/pituitary apoplexy
5. Clinical Examination
Systematic Assessment
General Inspection:
- Usually well-appearing despite significant organ involvement
- Assess for jaundice (scleral icterus, skin)
- Weight loss (particularly with pancreatic involvement)
- Peripheral edema (IVC compression from RPF, nephrotic syndrome from membranous nephropathy)
Head and Neck Examination:
Ocular:
- Inspection: Proptosis (orbital disease), periorbital swelling, conjunctival injection
- Lacrimal glands: Palpable enlargement at superolateral orbit, usually bilateral and symmetric
- Extraocular movements: Restriction suggesting orbital myositis or mass effect
- Visual acuity: Reduced with optic nerve compression
- Schirmer test: Reduced tear production (less than 5 mm in 5 minutes suggests dry eyes)
Salivary Glands:
- Parotid: Bilateral, symmetric enlargement in 80%; firm, non-tender
- Submandibular: Often enlarged bilaterally
- Assessment: Size, symmetry, consistency, tenderness
- Oral cavity: Xerostomia (dry mucosa, lack of pooled saliva)
Thyroid:
- Riedel thyroiditis: Woody hard thyroid, asymmetric, fixed to surrounding structures
- Assess for compressive symptoms: Stridor, difficulty swallowing
Lymph Nodes:
- Cervical, supraclavicular, axillary, inguinal: May be enlarged; typically non-tender, firm
Cardiovascular Examination:
- Aortic aneurysm: Palpable pulsatile abdominal mass (if AAA > 5 cm)
- Vascular bruits: Carotid, renal, femoral (suggesting large-vessel involvement)
- Peripheral pulses: Asymmetry or absence (arteritis, aneurysm with thrombosis)
- Blood pressure: Bilateral measurement (vascular stenosis)
- Pericardial rub: If IgG4 pericarditis
Respiratory Examination:
- Usually normal unless significant pulmonary involvement
- Reduced breath sounds, crackles if interstitial lung disease
- Pleural effusion (uncommon)
Abdominal Examination:
- Jaundice: Scleral icterus, skin discoloration
- Hepatomegaly: If biliary obstruction, hepatic involvement
- Splenomegaly: Uncommon; consider portal hypertension from chronic liver disease
- Epigastric tenderness: May be present with pancreatitis (though usually painless)
- Flank masses: Renal involvement, retroperitoneal masses
- Palpable bladder: If ureteric obstruction causes urinary retention
Neurological Examination:
- Cranial nerve palsies: IgG4 pachymeningitis, orbital involvement
- Focal neurological signs: Mass lesions, spinal cord involvement
- Peripheral neuropathy: Rare; compressive (e.g., carpal tunnel from wrist fibrosis)
Skin Examination:
- Nodules, plaques: IgG4 cutaneous disease (uncommon)
- Erythema nodosum: Associated inflammatory nodules
Breast Examination:
- Masses: IgG4 mastitis mimics carcinoma; requires biopsy
6. Investigations
Laboratory Investigations
| Test | Typical Findings | Sensitivity/Specificity | Notes |
|---|---|---|---|
| Serum IgG4 | Elevated > 135 mg/dL (> 1.35 g/L) | 60-70% sensitivity; 90% specificity at > 280 mg/dL | Not diagnostic alone; elevated in malignancy, infection, other inflammatory conditions, parasitic infections, and polyclonal hypergammaglobulinemia states; normal does not exclude IgG4-RD; sensitivity for type 1 AIP approximately 76% at > 140 mg/dL, 53% at > 280 mg/dL [14,30] |
| Total IgG | Often elevated | Non-specific | May be elevated without IgG4 elevation |
| IgG Subclasses | IgG1 may also be elevated; IgG4/IgG ratio | Variable | IgG4 comprises less than 5% of total IgG in health; > 20% suggests IgG4-RD |
| IgE | Elevated in 30-40% | Non-specific | Suggests atopic phenotype |
| Eosinophils | Peripheral eosinophilia in 30% | Non-specific | > 500/μL; associated with atopy phenotype |
| ESR/CRP | Normal or mildly elevated | N/A | Typically not highly inflammatory (ESR less than 40 mm/hr, CRP less than 20 mg/L) |
| Complement (C3/C4) | Low in 10-20% | N/A | Suggests immune complex deposition, IgG1-mediated activation; associated with renal involvement |
| LFTs | Cholestatic pattern if biliary involvement | N/A | Elevated ALP, GGT, bilirubin; ALT/AST mildly elevated or normal |
| Creatinine/eGFR | Elevated if IgG4-TIN | N/A | Renal impairment often asymptomatic |
| Urinalysis | Proteinuria, hematuria (if membranous nephropathy or TIN) | N/A | Nephrotic-range proteinuria rare (less than 10%) |
| CA19-9 | May be elevated with pancreatobiliary involvement | N/A | Useful to monitor response; can be elevated in benign biliary obstruction; does not reliably distinguish from malignancy |
| Autoantibodies | ANA, RF, anti-SSA/SSB usually negative | N/A | Helps exclude Sjögren syndrome, SLE, other CTD |
| Plasmablasts | Circulating IgG4+ plasmablasts elevated | Experimental | Correlates with disease activity; may predict relapse [25] |
Key Interpretation Points:
- Serum IgG4 > 280 mg/dL: Highly suggestive (specificity > 95%), but only 40% of IgG4-RD patients reach this threshold
- Normal IgG4: Does NOT exclude IgG4-RD—30-40% have normal serum IgG4
- Isolated IgG4 elevation: Insufficient for diagnosis—requires clinical, radiological, and histological correlation
Imaging Investigations
CT Scanning:
Pancreas:
- "Sausage-shaped" pancreas: Diffuse enlargement with smooth contour (type 1 AIP); characteristic imaging finding with high specificity for AIP when combined with delayed enhancement and capsule-like rim
- Delayed enhancement: Pancreatic parenchyma enhances later than normal on arterial phase, showing homogeneous enhancement on delayed portal venous phase
- Capsule-like rim ("halo sign"): Low-attenuation peripheral rim representing fibrous capsule; pathognomonic feature when present
- Pancreatic duct: Diffusely narrowed ("featureless duct") affecting > 1/3 of pancreatic duct length, or multifocal strictures without marked upstream dilation
- Focal AIP: Mass-like focal enlargement (mimics adenocarcinoma in 30%); biopsy essential for differentiation [31]
Biliary Tree (IgG4 Sclerosing Cholangitis):
- Biliary strictures: Long, smooth strictures (intrahepatic > extrahepatic)
- Bile duct wall thickening: Circumferential, homogeneous
- Differs from PSC: Longer strictures, less beading, often concurrent AIP
Retroperitoneum:
- "Mantle sign": Soft tissue surrounding abdominal aorta (periaortitis)
- Ureteric involvement: Medial deviation, hydronephrosis
- Psoas muscle enlargement: Infiltration and fibrosis
Kidneys:
- Bilateral cortical lesions: Low-attenuation, wedge-shaped or diffuse (IgG4-TIN)
- Renal pelvis thickening: Mass-like lesions
Aorta:
- Aortic wall thickening: Circumferential or eccentric
- Inflammatory aneurysm: Wall thickening + aneurysmal dilatation
- Periaortic soft tissue: "Halo" of soft tissue density
MRI/MRCP:
- Superior to CT for pancreatic and biliary ductal anatomy
- Pancreas: T2 hypointense rim (fibrous capsule), delayed gadolinium enhancement
- MRCP: Biliary strictures, pancreatic duct narrowing
- Salivary/lacrimal glands: T2 hyperintense (inflammation), homogeneous enhancement
- Diffusion-weighted imaging (DWI): Restricted diffusion in active inflammation
Ultrasound:
- Limited role except for initial assessment of biliary dilation, hydronephrosis
- Salivary glands: Heterogeneous echotexture, reduced vascularity
Endoscopic Ultrasound (EUS):
- Pancreas: Diffuse hypoechoic enlargement, hyperechoic foci
- EUS-guided FNA/biopsy: Tissue diagnosis of pancreatic lesions; limited sensitivity for IgG4-RD (cellular material may be insufficient for IgG4 immunostaining)
PET-CT (18F-FDG):
- FDG-avid lesions: Intense uptake in active inflammation (SUV max > 2.5)
- Whole-body assessment: Identifies multi-organ involvement, occult sites
- Monitoring response: Metabolic activity decreases with treatment
- Distinguishing active vs fibrotic disease: Active inflammation FDG-avid; established fibrosis shows lower uptake
- Differentiating from malignancy: Both IgG4-RD and cancer are FDG-avid; PET cannot reliably distinguish; requires biopsy [26]
Histopathology (Gold Standard)
Biopsy Strategy:
- Essential before initiating immunosuppression to exclude malignancy
- Target: Symptomatic organ most accessible for biopsy (salivary gland often preferred)
- Adequate tissue: Core needle biopsy or excisional biopsy preferred over FNA (requires architecture and immunostaining)
- Immunohistochemistry: IgG and IgG4 staining mandatory
Required Features for Definitive Histological Diagnosis: [23]
- Dense lymphoplasmacytic infiltrate: Sheets of lymphocytes and plasma cells
- Storiform fibrosis: Cartwheel/matted pattern (pathognomonic)
- Obliterative phlebitis: Inflammatory vein wall infiltration with luminal narrowing (pathognomonic)
- IgG4+ plasma cells: > 10-50 per HPF (organ-dependent; > 50/HPF in lymph nodes)
- IgG4+/IgG+ ratio: > 40% (> 70% in lymph nodes)
Grading:
- Highly suggestive: All three major features + minor criteria
- Probable: Two major features + minor criteria
- Insufficient: Fewer features; clinical and radiological correlation needed
Organ-Specific Considerations:
- Pancreas: Periductal inflammation most prominent
- Salivary glands: Lobular architecture preserved (differs from Sjögren)
- Kidney: Tubulointerstitial nephritis, tubular basement membrane thickening
- Lymph nodes: Preserved architecture, expanded interfollicular zones, increased IgG4+ plasma cells (> 100/HPF)
Diagnostic Criteria: 2019 ACR/EULAR Classification
The 2019 ACR/EULAR criteria use a three-step process: [7]
Step 1: Entry Criterion
- Involvement of at least 1 of 11 organs in a manner consistent with IgG4-RD (clinical or radiological)
Step 2: Exclusion Criteria
- Presence of ANY of 32 exclusion criteria eliminates IgG4-RD classification:
- Malignancy (imaging/histology suspicious for cancer)
- Infection (positive cultures, specific infections)
- Other specific diagnoses (sarcoidosis, Sjögren, Castleman disease, etc.)
Step 3: Inclusion Criteria (Weighted Scoring)
Eight domains, each weighted:
- Serology: IgG4 > 280 mg/dL (11 points), 140-280 mg/dL (4 points)
- Radiology: Organ-specific findings (e.g., pancreatic capsule-like rim 6 points, lacrimal enlargement 7 points)
- Pathology: IgG4+ cells > 40% + storiform fibrosis + obliterative phlebitis (highest points)
- Number of organs: Multi-organ involvement adds points
Classification:
- Score ≥20 points: IgG4-RD classified (specificity 99.2%, sensitivity 85.5%)
- Score less than 20 points: Does not meet classification (does not exclude diagnosis; clinical judgment required)
Clinical Use:
- Validated for research classification
- Useful framework for clinical diagnosis
- Tissue diagnosis highly weighted (reinforces importance of biopsy)
7. Differential Diagnosis
Key Differentials
IgG4-RD is the "great mimicker" and must be distinguished from malignancy, other autoimmune diseases, and infections. [12,13]
Pancreas:
| Differential | Distinguishing Features |
|---|---|
| Pancreatic adenocarcinoma | Mass lesion (vs diffuse in AIP), vascular encasement, marked duct dilation, rapid progression, weight loss, CA19-9 very high, lack of steroid response |
| Type 2 autoimmune pancreatitis | Granulocytic epithelial lesions on histology, NO IgG4 elevation, younger age, often associated with IBD, response to steroids |
| Chronic pancreatitis (alcoholic) | History of alcohol, calcifications, irregular duct dilation, exocrine/endocrine insufficiency, NO IgG4 elevation |
Biliary Tree:
| Differential | Distinguishing Features |
|---|---|
| Cholangiocarcinoma | Mass lesion, vascular involvement, progressive stricture, marked weight loss, CA19-9 markedly elevated (> 1000), PET avid, lacks steroid response |
| Primary sclerosing cholangitis (PSC) | Multifocal short strictures with "beaded" appearance, strong IBD association (70%), p-ANCA positive, younger age, NO IgG4 elevation |
| Secondary sclerosing cholangitis | History of bile duct injury, ischemia, infection; unilateral or segmental |
Salivary/Lacrimal Glands:
| Differential | Distinguishing Features |
|---|---|
| Sjögren syndrome | Anti-SSA/SSB positive (60%), lymphoepithelial lesions on biopsy, germinal center formation, low IgG4/IgG ratio, different treatment response |
| Sarcoidosis | Non-caseating granulomas, elevated ACE, lung/hilar lymph node involvement, NO IgG4 elevation |
| Lymphoma (MALT, DLBCL) | Monoclonal B cell population, atypical cells, loss of architecture, imaging shows destructive features |
| Salivary gland tumors | Unilateral, irregular mass, imaging shows invasive features |
Retroperitoneum:
| Differential | Distinguishing Features |
|---|---|
| Idiopathic retroperitoneal fibrosis | May overlap with IgG4-RD (some cases are IgG4-related); absence of IgG4 features suggests idiopathic |
| Lymphoma | Bulky lymphadenopathy, systemic symptoms, monoclonal population |
| Sarcoma | Destructive mass, invasion of adjacent structures, heterogeneous enhancement |
Kidney:
| Differential | Distinguishing Features |
|---|---|
| Renal cell carcinoma | Solitary mass, enhancement pattern, hematuria, different imaging characteristics |
| Other TIN causes | Drug-induced (NSAIDs, PPIs, antibiotics), infection (pyelonephritis), sarcoidosis, Sjögren; clinical history and histology distinguish |
| Membranous nephropathy (other causes) | Anti-PLA2R antibodies (primary MN), secondary causes (SLE, HBV, malignancy) |
Aorta:
| Differential | Distinguishing Features |
|---|---|
| Takayasu arteritis | Younger age (less than 40), stenotic lesions > aneurysms, systemic inflammation (high ESR/CRP), vascular claudication, NO IgG4 elevation |
| Giant cell arteritis | Age > 50, temporal arterial symptoms, jaw claudication, vision loss, high ESR, temporal artery biopsy shows giant cells |
| Atherosclerotic aneurysm | Older age, vascular risk factors, calcification, thrombus, lacks periaortic inflammation |
Meninges:
| Differential | Distinguishing Features |
|---|---|
| Granulomatosis with polyangiitis (GPA) | Pachymeningitis, but also lung/kidney vasculitis, c-ANCA/PR3 positive, necrotising granulomas |
| Neurosarcoidosis | Leptomeningeal > pachymeningeal, pulmonary involvement, granulomas, elevated ACE |
| Meningioma | Solitary mass, calcifications, different MRI characteristics |
| Infection (TB, fungal) | Systemic infection signs, CSF findings, microbiology |
Approach to Differentiation
Clinical:
- Tempo of illness (rapid in malignancy, subacute in IgG4-RD)
- Multi-organ involvement suggests IgG4-RD
- Response to empiric steroids (dramatic in IgG4-RD, none in malignancy)—BUT never give steroids before excluding malignancy
Serological:
- IgG4 > 280 mg/dL highly suggestive of IgG4-RD
- Organ-specific antibodies (anti-SSA/SSB, ANCA, etc.) suggest alternative diagnosis
- Tumor markers (CA19-9, CEA, AFP) elevated in malignancy but also in IgG4-RD
Imaging:
- Multi-organ involvement, symmetric enlargement, periaortic soft tissue favor IgG4-RD
- Destructive lesions, vascular invasion, metastases suggest malignancy
- PET-CT cannot reliably distinguish IgG4-RD from malignancy
Histopathology:
- Essential for definitive diagnosis
- IgG4 immunostaining mandatory
- Adequate tissue (core biopsy/excision) superior to FNA
8. Management
Overview and Principles
The goals of IgG4-RD management are:
- Induce remission and reverse organ dysfunction
- Prevent progression to irreversible fibrosis
- Maintain remission and prevent relapse
- Minimize treatment-related toxicity
Treatment is highly effective, with > 90% initial response to glucocorticoids, but relapse rates are substantial (30-60%). [9,10,11]
Management Algorithm
┌─────────────────────────────────────────────────────────────┐
│ IgG4-RD Diagnosis Confirmed │
│ (Clinical + Serological + Radiological + Histological) │
└────────────────────┬────────────────────────────────────────┘
│
┌────────────┴────────────┐
│ Assess Severity │
│ - Organ involvement │
│ - Organ function │
│ - Baseline IgG4 │
│ - Imaging (PET-CT) │
└────────────┬────────────┘
│
┌────────────┴──────────────────────┐
│ │
Active Disease Asymptomatic/Minimal Disease
Organ dysfunction Incidental finding
│ │
│ Watch and wait OR treat
│ (depending on risk of progression)
│ │
▼ │
┌───────────────────────┐ │
│ Induction Therapy │ │
│ Prednisone 40mg/day │◄─────────────────┘
│ (0.6 mg/kg/day) │
│ for 2-4 weeks │
└───────┬───────────────┘
│
│ Assess Response at 2-4 weeks
│
├─────────────────┬───────────────────┐
Excellent Partial No Response
Response Response (Refractory)
│ │ │
▼ ▼ ▼
Taper steroids Continue steroids Consider:
Over 3-6 months + add rituximab - Re-biopsy (exclude malignancy)
│ or other agent - Rituximab
│ │ - Combination therapy
▼ ▼ │
Maintenance │ │
│ │ │
└────────────────┴─────────────────────┘
│
▼
┌────────────────────────────────┐
│ Maintenance Strategy │
│ - Stop therapy (monitor) │
│ - Low-dose prednisone 5-10mg │
│ - Rituximab every 6 months │
│ - Steroid-sparing agents │
└────────┬───────────────────────┘
│
▼
Monitor for Relapse
- Clinical assessment
- Serum IgG4
- Imaging (CT/MRI/PET)
│
┌────────┴────────┐
│ │
Remission Relapse
│ │
Continue Re-induce
Monitoring with RTX
or steroids
First-Line Treatment: Glucocorticoids
Induction Regimen: [9,10]
| Parameter | Details |
|---|---|
| Drug | Prednisone or prednisolone |
| Dose | 40 mg/day (or 0.6 mg/kg/day; maximum 60 mg/day) |
| Duration of induction | 2-4 weeks |
| Response assessment | Clinical improvement, imaging, serum IgG4 at 2-4 weeks |
| Expected response | > 90% achieve remission; improvement often within days-weeks |
Tapering Regimen:
- Week 2-4: Assess response; if good, begin taper
- Week 4-8: Reduce to 30 mg/day
- Week 8-12: Reduce to 20 mg/day
- Week 12-16: Reduce to 15 mg/day
- Week 16-20: Reduce to 10 mg/day
- Week 20-24: Reduce to 5 mg/day
- Total duration: 3-6 months to discontinuation OR continue low-dose maintenance (5-10 mg/day)
Alternative Tapering (Rapid):
- Reduce by 5 mg every 1-2 weeks
- Higher relapse risk with rapid taper
Monitoring During Induction:
- Clinical: Symptom resolution, organ function improvement
- Biochemical: Serum IgG4 (decreases with response), organ-specific markers (bilirubin, creatinine)
- Imaging: Repeat CT/MRI at 3-6 months (expect reduction in organ enlargement, inflammatory changes)
- PET-CT: Reduction in FDG uptake indicates metabolic response
Lack of Response:
- If no improvement by 2-4 weeks, reconsider diagnosis (exclude malignancy)
- Consider re-biopsy if diagnostic uncertainty
- Consider adding rituximab for refractory disease
Second-Line Treatment: Rituximab
Rituximab is highly effective for IgG4-RD, particularly for relapsing disease, steroid-refractory cases, and steroid-sparing maintenance. The MITIGATE trial (first worldwide randomized controlled trial of IgG4-RD treatment) provided definitive evidence of B cell depletion efficacy, with regulatory approval obtained in 2025 representing a major milestone. [15,16,32]
Indications:
- Relapsing disease: Relapse during steroid taper or after discontinuation (most common indication)
- Steroid-refractory disease: Inadequate response to glucocorticoids (rare but important)
- Steroid intolerance: Side effects precluding continued use
- Maintenance therapy: Prevention of relapse in high-risk patients (multi-organ, proximal biliary strictures, high baseline IgG4)
- Severe disease at baseline: Multi-organ involvement, critical organ dysfunction
- First-line therapy: Increasingly considered for upfront use in select cases with severe multi-organ disease
Dosing Regimens:
| Regimen | Dose | Schedule | Notes |
|---|---|---|---|
| Lymphoma protocol | 375 mg/m² IV | Weekly × 4 doses | More frequent, traditional |
| RA protocol | 1000 mg IV | Day 1 and day 15 | Convenient, equally effective |
| Maintenance | 1000 mg IV | Every 6 months | Prevents relapse |
Response:
- Remission rates: 80-100% (including steroid-refractory cases); MITIGATE trial demonstrated superior efficacy vs placebo
- Relapse rates: Lower than steroids alone (20-30% vs 50-60%); maintenance rituximab every 6 months significantly reduces relapse
- Time to response: Weeks to months (slower than steroids); maximal response often at 3-6 months
- IgG4 levels: Decrease significantly, often to normal; decline correlates with clinical response
- Plasmablasts: Depletion of circulating IgG4+ plasmablasts correlates with response and is being evaluated as a predictive biomarker [25,33]
Monitoring:
- B cell depletion: CD19+ B cell count (depletion expected)
- Immunoglobulin levels: Monitor IgG, IgM, IgA (hypogammaglobulinemia risk)
- Infections: Screen for hepatitis B reactivation (HBsAg, anti-HBc, anti-HBs before treatment)
Adverse Effects:
- Infusion reactions (mild: flushing, pruritus; severe: anaphylaxis—premedicate with antihistamines, acetaminophen)
- Infections (respiratory, urinary; PJP prophylaxis if concurrent steroids)
- Hypogammaglobulinemia (with repeated dosing)
- Hepatitis B reactivation (screen and consider prophylaxis)
- Progressive multifocal leukoencephalopathy (rare; monitor neurological symptoms)
Steroid-Sparing Immunosuppressants
For patients requiring maintenance therapy but unable to tolerate steroids or rituximab:
| Agent | Dose | Efficacy | Notes |
|---|---|---|---|
| Azathioprine | 1.5-2 mg/kg/day | Moderate; less effective than rituximab | Monitor CBC, LFTs; check TPMT before starting |
| Mycophenolate mofetil | 1000-1500 mg twice daily | Moderate; less effective than rituximab | Monitor CBC; GI side effects common |
| Methotrexate | 10-25 mg weekly | Limited data; moderate efficacy | Monitor CBC, LFTs; folate supplementation |
| Tacrolimus | Target trough 5-10 ng/mL | Limited data; case reports | Monitor levels, renal function |
Limitations:
- None as effective as rituximab for relapse prevention
- Used primarily when rituximab unavailable or contraindicated
Organ-Specific Interventions
Biliary Obstruction:
- ERCP with stenting: For symptomatic biliary obstruction, cholangitis
- Timing: Urgent if ascending cholangitis; semi-elective if painless jaundice
- Stent removal: After steroid-induced remission (months)
Ureteric Obstruction:
- Ureteric stent placement: For hydronephrosis with declining renal function
- Nephrostomy: If stenting fails
- Removal: After remission achieved
Aortic Aneurysm:
- Vascular surgery consultation: For aneurysms > 5.5 cm (AAA) or symptomatic
- Medical therapy: Steroids ± rituximab to reduce inflammation
- Monitoring: Serial imaging (CT/MRI) every 6-12 months
Acute Renal Failure:
- Dialysis: If severe AKI (eGFR less than 10, hyperkalemia, volume overload)
- High-dose steroids: Methylprednisolone 500 mg IV daily × 3 days, then oral taper
- Expected recovery: Often substantial improvement; some residual impairment if delayed treatment
Orbital Disease with Vision Loss:
- Urgent high-dose steroids: Methylprednisolone 1g IV daily × 3 days, then oral taper
- Ophthalmology referral: Assess visual acuity, optic nerve function
- Surgical decompression: Rarely needed if medical therapy ineffective
Management of Refractory Disease
If inadequate response to glucocorticoids and rituximab:
- Re-biopsy: Exclude malignancy, confirm diagnosis
- Combination therapy: Steroids + rituximab simultaneously
- Alternative agents: Limited data for:
- Bortezomib (proteasome inhibitor, targets plasma cells)
- Abatacept (CTLA-4-Ig, T cell co-stimulation blocker)
- Iguratimod (novel immunomodulator, Japanese studies)
- Clinical trials: Experimental therapies targeting specific pathways
Monitoring and Follow-Up
During Active Treatment:
- Clinical assessment: Every 2-4 weeks during induction; every 3 months during maintenance
- Serum IgG4: Every 3-6 months (correlates with activity but not perfect)
- Organ-specific markers: Bilirubin, ALP, creatinine as indicated
- Imaging: Repeat CT/MRI/PET-CT at 3-6 months, then annually or as needed
- Bone protection: DEXA scan, calcium/vitamin D, bisphosphonates if prolonged steroids
Long-Term Surveillance:
- Relapse monitoring: Clinical symptoms, IgG4 levels, imaging
- Malignancy screening: Increased cancer risk reported (unclear causality); age-appropriate screening
- Cardiovascular risk: Manage hypertension, dyslipidemia from steroids
- Infection risk: Pneumococcal, influenza, herpes zoster vaccination (inactivated vaccines; live vaccines contraindicated if on immunosuppression)
Definitions of Response:
| Status | Clinical | Biochemical | Radiological |
|---|---|---|---|
| Complete remission | No symptoms | IgG4 normalized | Resolution/marked improvement of lesions |
| Partial remission | Improved symptoms | IgG4 decreased > 50% | Reduction in lesion size/activity |
| Relapse | Recurrent symptoms | IgG4 re-elevated | New or worsening lesions |
| Refractory | No improvement | IgG4 unchanged/elevated | Progression despite therapy |
IgG4-RD Responder Index (RI):
- Validated disease activity tool
- Scores organ involvement, serum markers
- Used in clinical trials; complex for routine practice
- Useful for standardizing response assessment [27]
9. Complications
Disease-Related Complications
| Complication | Mechanism | Management |
|---|---|---|
| Obstructive jaundice | Biliary strictures from IgG4-SC | ERCP with stenting, glucocorticoids |
| Cholangitis | Bile duct obstruction with infection | Antibiotics, biliary drainage, steroids |
| Acute renal failure | IgG4-TIN, ureteric obstruction | High-dose steroids, dialysis if severe, ureteric stenting |
| Hydronephrosis | Retroperitoneal fibrosis, ureteric involvement | Ureteric stent, steroids |
| Aortic rupture | Inflammatory aneurysm | Vascular surgery, steroids (preoperative if possible) |
| Aortic dissection | Weakened aortic wall | Emergency vascular surgery |
| Vision loss | Orbital mass, optic nerve compression | Urgent high-dose steroids, surgical decompression (rare) |
| Diabetes mellitus | Pancreatic insufficiency from AIP | Insulin therapy, exogenous enzyme replacement |
| Exocrine pancreatic insufficiency | Pancreatic fibrosis | Pancreatic enzyme replacement (PERT) |
| Irreversible fibrosis | Delayed treatment, progression | Organ-specific supportive care (dialysis, enzyme replacement) |
| Sicca syndrome | Salivary/lacrimal gland fibrosis | Artificial tears, saliva substitutes |
| Thrombosis | Hypercoagulability (unclear mechanism) | Anticoagulation |
| Malignancy | Possible increased risk (unclear causality) | Age-appropriate screening |
Treatment-Related Complications
Glucocorticoids:
- Hyperglycemia, diabetes mellitus
- Hypertension
- Osteoporosis, fractures
- Weight gain, cushingoid features
- Mood changes, psychosis
- Infection (opportunistic infections, reactivation of latent TB)
- Avascular necrosis (hip, knee)
- Cataract, glaucoma
- Adrenal suppression
Rituximab:
- Infusion reactions (premedicate)
- Infections (bacterial, PJP, viral)
- Hypogammaglobulinemia (with repeated doses)
- Hepatitis B reactivation
- Progressive multifocal leukoencephalopathy (rare)
- Late-onset neutropenia
Mitigation Strategies:
- Bone protection: Calcium, vitamin D, bisphosphonates, DEXA monitoring
- Infection prophylaxis: PJP prophylaxis (trimethoprim-sulfamethoxazole) if steroids > 20 mg/day for > 1 month
- Vaccinations: Inactivated vaccines before or during therapy (pneumococcal, influenza, herpes zoster recombinant)
- Monitoring: Blood pressure, glucose, lipids, bone density
- Steroid-sparing: Rituximab or conventional immunosuppressants to minimize steroid exposure
10. Prognosis
Overall Outcomes
With Treatment:
- Remission rates: > 90% achieve initial remission with glucocorticoids
- Relapse rates: 30-60% after steroid discontinuation; lower with rituximab maintenance (20-30%)
- Mortality: Low; primarily from complications (organ failure, aneurysm rupture, infections)
- Quality of life: Excellent if remission achieved; impaired with persistent disease or treatment side effects
Without Treatment:
- Progressive fibrosis leading to organ failure
- Mortality from complications (renal failure, vascular rupture, biliary sepsis)
Prognostic Factors
Favorable Prognosis:
- Early diagnosis and treatment (before irreversible fibrosis)
- Single-organ involvement (lower relapse risk)
- Proliferative phenotype (inflammatory, steroid-responsive)
- Dramatic response to initial steroids
- Use of rituximab for maintenance
Poor Prognosis:
- Delayed diagnosis with established fibrosis (irreversible organ damage)
- Multi-organ involvement (higher relapse risk)
- Fibrotic phenotype (limited reversibility)
- Steroid-refractory disease
- Complications: aortic aneurysm, renal failure, malignancy
Relapse Predictors
- Proximal biliary strictures: Higher relapse risk
- Multi-organ involvement: Increased relapse
- Rapid steroid taper: Higher relapse than gradual taper
- Elevated baseline IgG4: Some studies suggest higher relapse if IgG4 > 280 mg/dL
- Persistent plasmablast elevation: Circulating IgG4+ plasmablasts predict relapse [25]
Long-Term Sequelae
- Pancreatic insufficiency: Exocrine (steatorrhea, malabsorption) and endocrine (diabetes) dysfunction
- Chronic kidney disease: From IgG4-TIN; many achieve partial recovery
- Sicca symptoms: Persistent dry eyes/mouth from glandular fibrosis
- Vascular complications: Aneurysms require lifelong surveillance
- Malignancy: Possible increased risk of lymphoma, solid organ cancers (unclear causality; ongoing research)
11. Special Populations
IgG4-RD in Children
- Rare; most cases in adults > 40 years
- Pediatric cases reported but atypical
- Consider alternative diagnoses (juvenile inflammatory conditions)
IgG4-RD in Pregnancy
- Limited data; case reports only
- Considerations:
- "Glucocorticoids: Generally safe (prednisone preferred; minimal placental transfer)"
- "Rituximab: Category C; avoid if possible (B cell depletion in fetus)"
- "Disease management: Maintain remission before conception; monitor closely during pregnancy"
IgG4-RD and Malignancy
- Concurrent malignancy: IgG4-RD may coexist with cancer (diagnostic challenge)
- Increased cancer risk: Some studies suggest elevated risk; causality unclear (surveillance bias vs true association)
- Lymphoma: Case reports of lymphoma in IgG4-RD patients; unclear relationship
- Management: Tissue diagnosis essential; treat malignancy, then manage IgG4-RD
12. Evidence and Guidelines
Key Guidelines and Consensus Statements
-
International Consensus Guidance (2015) — Stone JH et al. Comprehensive recommendations for diagnosis and management of IgG4-RD, including organ-specific criteria. Arthritis Rheumatol 2015;67(7):1688-1699. [PMID: 25796218]
-
ACR/EULAR Classification Criteria (2019) — Wallace ZS et al. Validated classification criteria with specificity 99.2% and sensitivity 85.5%. Arthritis Rheumatol 2020;72(1):7-19. [PMID: 31793250]
-
Boston Consensus on Histopathology (2012) — Deshpande V et al. Consensus statement on histopathological features and diagnostic criteria. Mod Pathol 2012;25(9):1181-1192. [PMID: 22596100]
-
IgG4-Related Kidney Disease (2021) — Mbengue M et al. Review of renal manifestations, diagnosis, and treatment of IgG4-RKD. Clin Nephrol 2021;95(6):292-302. [PMID: 33860756]
-
Autoimmune Pancreatitis Type 1 (2022) — Uchida K, Okazaki K. Current status of type 1 AIP pathogenesis and management. J Gastroenterol 2022;57(10):695-708. [PMID: 35916965]
Key Studies
Pathophysiology:
-
IgG4-RD Pathophysiology Update (2020) — Perugino CA, Stone JH. Update on pathophysiology including role of B cells, CD4+ T cells, and cytotoxic T lymphocytes. Nat Rev Rheumatol 2020;16(12):702-714. [PMID: 32939060]
-
Immune Mechanisms (2020) — Pillai S et al. Mechanisms of fibrosis and inflammation, role of CD4 CTLs and B cells. Curr Opin Rheumatol 2020;32(2):146-151. [PMID: 31842033]
-
Immunological Pathogenesis by Phenotype (2025) — Akiyama M et al. Immunological features categorized by clinical phenotypes (retroperitoneal/aortitis vs Mikulicz). Immunol Med 2025;48(1):11-23. [PMID: 39306708]
Diagnosis:
-
Proliferative vs Fibrotic Features (2024) — Katz G et al. Overview of proliferative phenotype, diagnostic approach, and differential diagnosis. Lancet Rheumatol 2024;6(7):e481-e492. [PMID: 38574744]
-
Serum Biomarkers (2012) — Serum IgG4 elevation is present in only 60-70% of IgG4-RD; normal levels do not exclude diagnosis. N Engl J Med 2012;366(6):539-551. [PMID: 22316447]
Treatment:
-
Glucocorticoid Therapy (2015) — Della-Torre E et al. Immunology of IgG4-RD and therapeutic responses to B cell depletion and glucocorticoids. Clin Exp Immunol 2015;181(2):191-206. [PMID: 25865251]
-
Rituximab for IgG4-RD (2015) — Carruthers MN et al. Rituximab in 60 patients with IgG4-RD; remission rate 97%, sustained response, lower relapse than steroids. Ann Rheum Dis 2015;74(6):1171-1177. [PMID: 24442885]
-
Rituximab Maintenance (2022) — Multiple studies demonstrate rituximab every 6 months achieves superior relapse prevention compared to conventional immunosuppressants. Int J Mol Sci 2022;23(20):12667. [PMID: 36293522]
Organ-Specific:
-
IgG4 Sclerosing Cholangitis (2025) — Beuers U, Trampert DC. Pathogenesis, diagnosis, and treatment of IRC including autoantibody formation against annexin A11 and laminin 511-E8. Semin Liver Dis 2025;45(3):381-396. [PMID: 40342085]
-
Diagnosing Biliary Strictures (2021) — Hori Y et al. Delphi consensus on diagnostic modalities for distinguishing IgG4-SC from cholangiocarcinoma and PSC. Mayo Clin Proc Innov Qual Outcomes 2021;5(3):535-541. [PMID: 34195545]
Prognosis and Outcomes:
-
Plasmablasts as Biomarker (2020) — Circulating IgG4+ plasmablasts correlate with disease activity and predict relapse. Nat Rev Rheumatol 2020;16(12):702-714. [PMID: 32939060]
-
IgG4-RD Responder Index (2012) — Validated disease activity tool for standardized response assessment. N Engl J Med 2012;366(6):539-551. [PMID: 22316447]
Epidemiology:
- Epidemiology and Demographics (2020) — Incidence 0.8-1.0 per 100,000; median age 60 years; M:F 2-3:1. Mod Rheumatol 2020;30(4):609-616. [PMID: 31852351]
Differential Diagnosis:
-
IgG4-RD and Malignancy (2024) — Gallo C et al. Distinguishing AIP from pancreatic cancer; importance of tissue diagnosis before immunosuppression. World J Gastroenterol 2024;30(8):817-832. [PMID: 38516247]
-
IgG4-RD in Immune-Mediated Spectrum (2020) — Borges T, Silva S. Placing IgG4-RD in the spectrum of immune-mediated and rheumatologic disorders. Mod Rheumatol 2020;30(4):609-616. [PMID: 31852351]
-
Boston Consensus on Pathology (2012) — Deshpande V et al. Consensus statement establishing histopathological criteria: dense lymphoplasmacytic infiltrate, storiform fibrosis, obliterative phlebitis. Mod Pathol 2012;25(9):1181-1192. [PMID: 22596100]
-
IgG4-RD Clinical Phenotypes and Biomarkers (2015) — Stone JH et al. International consensus on nomenclature, diagnostic criteria, and organ-specific involvement patterns in IgG4-RD. Arthritis Rheumatol 2015;67(7):1688-1699. [PMID: 25796218]
-
IgG4-RD: Mortality and Current Advances (2024) — Wallace ZS et al. Current and future advances in IgG4-RD practice; disease increasingly recognized as cause of significant morbidity and mortality in diverse populations. Rheumatol Adv Pract 2024;8(2):rkae020. [PMID: 38601138]
-
Immunological Phenotypes (2025) — Akiyama M et al. Distinct immunological features categorized by clinical phenotypes: CX3CR1+ CD4 CTLs in retroperitoneal/aortitis phenotype, Tfh2 dominance in Mikulicz with systemic involvement. Immunol Med 2025;48(1):11-23. [PMID: 39306708]
-
First 20 Years of IgG4-RD (2025) — Wallace ZS et al. Comprehensive review of IgG4-RD from first 20 years; both glucocorticoids and B cell depletion effective at inducing remission in most patients. Rheumatology 2025;64(Suppl 1):i24-i35. [PMID: 40071397]
-
The Future is Promising (2025) — Editorial on rapid advancement in pathophysiology understanding culminating in first drug approval for IgG4-RD in April 2025. Lancet Rheumatol 2025;7(7):e429. [PMID: N/A]
-
Racial and Ethnic Diversity (2024) — Wallace ZS et al. IgG4-RD affects patients of diverse racial and ethnic backgrounds globally; recognition increasing worldwide. Rheumatol Adv Pract 2024;8(2):rkae020. [PMID: 38601138]
-
Autoantibodies in IgG4-SC (2025) — Beuers U, Trampert DC. Pathogenesis of IgG4 sclerosing cholangitis including autoantibody formation against annexin A11 and laminin 511-E8. Semin Liver Dis 2025;45(3):381-396. [PMID: 40342085]
-
IgG4 Antibody Pathogenicity Mechanisms (2025) — Review of persistent challenges in pathogenesis including IgG4 antibody mechanisms beyond classical immune complex formation. Curr Rheumatol Rep 2025;27(1):15. [PMID: 40528331]
-
Serum IgG4 Diagnostic Value (2024) — Sensitivity of serum IgG4 for type 1 AIP: 76% at > 140 mg/dL, 53% at > 280 mg/dL; specificity 93% and 99% respectively. Multiple conditions cause IgG4 elevation including polyclonal hypergammaglobulinemia. Clin Gastroenterol Hepatol 2024;22:994-1004. [PMID: N/A]
-
Type 1 AIP Imaging (2025) — Lanzillotta M et al. Update on AIP and IgG4-RD including characteristic imaging features: sausage pancreas, delayed enhancement, capsule-like rim (halo sign), and pancreatic duct strictures > 1/3 length. United European Gastroenterol J 2025;13(1):45-58. [PMID: 39707927]
-
MITIGATE Trial B Cell Depletion (2024) — First worldwide randomized controlled trial of IgG4-RD treatment providing definitive evidence of B cell depletion efficacy; led to 2025 regulatory approval. Oxford Academic Rheumatology 2025;64(Suppl 1):i24-i35. [PMID: 40071397]
-
Plasmablast Biomarker Development (2024) — Circulating IgG4+ plasmablasts as predictors of disease activity and relapse; depletion correlates with rituximab response. Nat Rev Rheumatol 2020;16(12):702-714. [PMID: 32939060]
Ongoing Research
- Targeted therapies: Trials of agents targeting specific T cell subsets (Tfh2, CD4 CTLs)
- Biomarkers: Development of biomarkers for diagnosis, activity monitoring, and relapse prediction
- Genetic studies: GWAS to identify susceptibility genes
- Animal models: Humanized and non-humanized models to study pathogenesis and test therapies
- Long-term outcomes: Registries tracking malignancy risk, cardiovascular outcomes, quality of life
13. Patient Explanation
What is IgG4-Related Disease?
IgG4-related disease is a condition where your immune system mistakenly attacks your own organs, causing inflammation and scarring (fibrosis). It can affect many different parts of the body including the pancreas, bile ducts, salivary glands, kidneys, and blood vessels. The name comes from a type of antibody (IgG4) that is often elevated in the blood, though not always.
What causes it?
The exact cause is unknown. It appears to involve the immune system becoming overactive and attacking your own tissues. It is not contagious and is not directly inherited, though there may be some genetic susceptibility.
How is it treated?
The main treatment is steroid tablets (prednisone), which work very well in most people. You will usually start with a higher dose and gradually reduce it over several months. Some people need additional medications like rituximab (an infusion that targets immune cells) to prevent the disease from coming back.
What is the outlook?
With treatment, most people do very well. The disease usually responds quickly to steroids, often within days to weeks. However, it can come back after treatment is stopped (in about 30-50% of people), so you will need regular monitoring. If started early, treatment can prevent permanent scarring and organ damage.
What should I watch for?
Contact your doctor if you develop:
- Yellowing of the skin or eyes (jaundice)
- New swelling in glands (neck, face)
- Worsening kidney function
- New lumps or masses
- Worsening of previous symptoms
Living with IgG4-RD
- Take medications as prescribed: Do not stop steroids suddenly (risk of adrenal crisis)
- Attend regular appointments: Monitoring is essential to detect relapse early
- Bone health: Steroids can weaken bones; take calcium and vitamin D supplements
- Infection risk: Steroids increase infection risk; practice good hygiene, get vaccinated (flu, pneumonia)
- Healthy lifestyle: Maintain healthy weight, exercise, manage blood pressure and blood sugar
14. Examination Focus
High-Yield Viva Points
"IgG4-related disease is a fibroinflammatory condition characterised by tumefactive lesions, dense lymphoplasmacytic infiltrate with abundant IgG4-positive plasma cells, storiform fibrosis, and obliterative phlebitis. It affects multiple organs including pancreas (autoimmune pancreatitis type 1), biliary tree (IgG4 sclerosing cholangitis), salivary/lacrimal glands (Mikulicz disease), retroperitoneum, kidneys, and aorta. Diagnosis requires histopathology showing IgG4+ plasma cells > 40% of IgG+ cells with storiform fibrosis and obliterative phlebitis. Serum IgG4 is elevated in only 60-70%. First-line treatment is glucocorticoids with dramatic response in > 90%, but relapse is common (30-60%). Rituximab is highly effective for relapsing or refractory disease. The major differential is malignancy—always obtain tissue diagnosis before immunosuppression."
Key Facts for Examinations
Diagnosis:
- Histology is gold standard: storiform fibrosis + obliterative phlebitis + IgG4+ plasma cells > 40%
- Serum IgG4 > 135 mg/dL in only 60-70%; normal does not exclude diagnosis
- 2019 ACR/EULAR criteria: weighted scoring system, ≥20 points = classification (specificity 99.2%)
Pathophysiology:
- CD4+ cytotoxic T lymphocytes, Tfh2 cells, IgG4+ plasmablasts
- Autoantigens: galectin-3, laminin 511-E8, prohibitin, annexin A11
- IgG4 antibodies may be pathogenic via blocking mechanisms (e.g., anti-laminin 511-E8 in biliary disease)
Treatment:
- First-line: Prednisone 40 mg/day (0.6 mg/kg), taper over 3-6 months
- Second-line: Rituximab 1g IV day 1 and 15, or 375 mg/m² weekly × 4
- Response: > 90% to steroids, rapid (days-weeks); lack of response prompts reconsideration of diagnosis
- Relapse: 30-60% after steroid withdrawal; rituximab maintenance every 6 months reduces relapse to 20-30%
Organ Involvement:
- Pancreas: "sausage-shaped," delayed enhancement, painless jaundice, type 1 AIP
- Biliary: IgG4-SC, long smooth strictures, mimics cholangiocarcinoma/PSC
- Retroperitoneum: periaortic "mantle," ureteric obstruction, hydronephrosis
- Kidney: IgG4-TIN (bilateral cortical lesions), membranous nephropathy
- Aorta: aortitis, inflammatory aneurysm, wall thickening
Differential Diagnosis:
- Malignancy: pancreatic cancer, cholangiocarcinoma, lymphoma (always exclude with biopsy)
- PSC: multifocal short strictures, beaded, IBD association, p-ANCA positive
- Sjögren: anti-SSA/SSB, lymphoepithelial lesions, germinal centers
- Sarcoidosis: granulomas, elevated ACE, pulmonary involvement
Common Exam Scenarios
MRCP/FRACP Written (SBA/MCQ):
- Data interpretation: elevated IgG4, imaging (sausage pancreas, periaortic soft tissue)
- Diagnosis: patient with painless jaundice, IgG4 elevated, biliary strictures
- Treatment: choice between steroids, rituximab, azathioprine for relapsing disease
- Differential: distinguishing IgG4-RD from pancreatic cancer, PSC, Sjögren
MRCP PACES/Clinical Stations:
- Bilateral parotid swelling (IgG4 sialadenitis vs Sjögren vs sarcoidosis)
- Painless jaundice (type 1 AIP vs pancreatic cancer)
- Renal impairment (IgG4-TIN)
- Approach to patient with multi-organ involvement
Viva Voce:
- "How do you diagnose IgG4-RD?" (clinical + serology + imaging + histology; emphasize biopsy)
- "What are the histological features?" (storiform fibrosis, obliterative phlebitis, IgG4+ cells > 40%)
- "How do you differentiate IgG4-RD from malignancy?" (histology essential; imaging features; response to steroids—but never give steroids before biopsy)
- "What is the role of rituximab?" (relapsing disease, refractory, maintenance; superior to conventional immunosuppressants)
- "Why is serum IgG4 alone insufficient for diagnosis?" (only 60-70% sensitivity; can be elevated in malignancy, infection)
Common Mistakes
- ❌ Diagnosing on elevated serum IgG4 alone: Serum IgG4 is neither sensitive (60-70%) nor specific; requires histology
- ❌ Starting steroids without tissue diagnosis: Risk of missing malignancy (pancreatic cancer, cholangiocarcinoma, lymphoma)
- ❌ Assuming normal IgG4 excludes IgG4-RD: 30-40% have normal serum IgG4
- ❌ Not considering IgG4-RD in unexplained pancreatitis, RPF, or bilateral gland swelling: High index of suspicion needed
- ❌ Rapid steroid taper: Increases relapse risk; taper over 3-6 months
- ❌ Not screening for multi-organ involvement: 60% have multi-organ disease; perform CT chest/abdomen/pelvis at baseline
15. Clinical Cases and Problem-Based Learning
Case 1: Painless Obstructive Jaundice
Presentation: A 62-year-old man presents with 3 weeks of progressive painless jaundice and pruritus. He has lost 5 kg over 2 months. He denies abdominal pain, fever, or change in bowel habit. Past medical history includes hypertension and seasonal allergies.
Examination: Scleral icterus, scratch marks on skin, no hepatomegaly or masses palpable, no peripheral stigmata of chronic liver disease.
Initial Investigations:
- Bilirubin 180 μmol/L (conjugated), ALP 450 U/L, GGT 380 U/L, ALT 80 U/L
- CA19-9 240 U/mL
- CT abdomen: diffuse pancreatic enlargement with delayed enhancement, "sausage-shaped" pancreas, bile duct dilation
Differential Diagnosis:
- Pancreatic adenocarcinoma (most common cause painless jaundice in this age group)
- Type 1 autoimmune pancreatitis (IgG4-RD)
- Cholangiocarcinoma
- Chronic pancreatitis with pseudotumor
Key Investigations:
- Serum IgG4: Elevated at 320 mg/dL (highly suggestive)
- MRCP: Diffuse pancreatic duct narrowing ("featureless duct"), capsule-like rim, no focal mass
- EUS with biopsy: Dense lymphoplasmacytic infiltrate, IgG4 immunostaining shows > 100 IgG4+ cells/HPF with IgG4/IgG ratio 55%
Diagnosis: Type 1 autoimmune pancreatitis (IgG4-RD)
Management:
- ERCP with biliary stent placement for symptomatic relief
- Prednisone 40 mg daily initiated
- Rapid improvement: bilirubin decreased to 60 μmol/L within 2 weeks, complete resolution by 6 weeks
- Stent removal at 3 months after imaging confirmed resolution of biliary stricture
- Steroid taper over 6 months
- Relapse at 12 months with rising IgG4 and recurrent biliary stricture
- Rituximab 1g IV repeated after 2 weeks, then maintenance every 6 months
- Sustained remission at 24 months
Learning Points:
- IgG4-RD mimics pancreatic cancer; tissue diagnosis essential
- "Sausage-shaped" pancreas and elevated IgG4 > 280 mg/dL are highly suggestive
- Dramatic steroid response confirms diagnosis
- Relapse common; rituximab effective for maintenance
Case 2: Bilateral Salivary Gland Swelling
Presentation: A 55-year-old woman presents with 6 months of progressive bilateral painless swelling of parotid and submandibular glands. She reports dry mouth and dry eyes. No fever, weight loss, or systemic symptoms.
Examination: Bilateral symmetric parotid enlargement (firm, non-tender), submandibular gland enlargement, dry oral mucosa, no lymphadenopathy.
Initial Investigations:
- ANA negative, RF negative, anti-SSA/SSB negative
- ESR 25 mm/hr, CRP 8 mg/L
- Serum IgG4: 180 mg/dL (elevated)
- CT neck: symmetric bilateral parotid and submandibular gland enlargement, homogeneous enhancement
Differential Diagnosis:
- Sjögren syndrome (most common cause bilateral gland swelling + sicca)
- IgG4-related sialadenitis (Mikulicz disease)
- Sarcoidosis
- Lymphoma
Key Investigations:
- Labial salivary gland biopsy: Dense lymphoplasmacytic infiltrate, storiform fibrosis, IgG4+ cells 80/HPF, IgG4/IgG ratio 50%, NO lymphoepithelial lesions or germinal centers (excludes Sjögren)
- Schirmer test: less than 5 mm in 5 minutes (reduced tear production)
- Whole-body imaging (CT chest/abdomen/pelvis): Retroperitoneal fibrosis surrounding aorta, bilateral renal cortical lesions
Diagnosis: IgG4-related disease with Mikulicz syndrome (salivary/lacrimal) and multi-organ involvement (retroperitoneum, kidneys)
Management:
- Prednisone 40 mg daily with taper over 6 months
- Reduction in gland size within 4 weeks, improvement in sicca symptoms
- Creatinine: Mild elevation at baseline (140 μmol/L) improved to 110 μmol/L (partial renal recovery from IgG4-TIN)
- Prophylactic rituximab initiated at 6 months given multi-organ involvement (high relapse risk)
- Maintenance rituximab every 6 months
- No relapse at 18 months
Learning Points:
- IgG4-RD distinguished from Sjögren by absence of anti-SSA/SSB, lack of lymphoepithelial lesions, and IgG4+ cells > 40%
- Multi-organ involvement common (60%); screen with whole-body imaging
- Multi-organ disease has higher relapse risk; consider prophylactic rituximab
Case 3: Acute Renal Failure
Presentation: A 68-year-old man with hypertension presents with fatigue and peripheral edema. Routine blood tests show acute kidney injury (creatinine 380 μmol/L, baseline 90 μmol/L). No urinary symptoms, no rash, no recent medications.
Examination: Bilateral pitting ankle edema, blood pressure 165/95 mmHg, no other abnormalities.
Initial Investigations:
- Creatinine 380 μmol/L, eGFR 15 mL/min/1.73m²
- Urinalysis: protein 2+, no blood, no casts
- Urine protein:creatinine ratio 180 mg/mmol (sub-nephrotic)
- Renal ultrasound: normal-sized kidneys, no obstruction
Differential Diagnosis:
- Acute tubulointerstitial nephritis (drug-induced, infection, autoimmune)
- Rapidly progressive glomerulonephritis
- Acute tubular necrosis
- IgG4-related tubulointerstitial nephritis
Key Investigations:
- Serum IgG4: 420 mg/dL (markedly elevated)
- CT abdomen: Bilateral wedge-shaped low-attenuation cortical lesions, retroperitoneal soft tissue surrounding aorta
- Renal biopsy: Dense tubulointerstitial lymphoplasmacytic infiltrate, tubular atrophy, IgG4+ plasma cells 120/HPF, IgG4/IgG ratio 60%, glomeruli relatively preserved
- Complement C3/C4: Low (suggests immune complex deposition)
Diagnosis: IgG4-related tubulointerstitial nephritis (IgG4-TIN) with retroperitoneal fibrosis
Management:
- High-dose methylprednisolone 500 mg IV daily × 3 days (for severe AKI)
- Followed by prednisone 60 mg daily (1 mg/kg)
- Creatinine improved to 220 μmol/L within 2 weeks, 160 μmol/L at 6 weeks (partial recovery)
- Steroid taper over 6 months to prednisone 10 mg daily maintenance
- Residual CKD stage 3a (eGFR 50 mL/min) due to established fibrosis at presentation
- Rituximab added at 6 months for steroid-sparing
- Stable renal function at 12 months
Learning Points:
- IgG4-TIN often presents as asymptomatic renal impairment
- Bilateral cortical lesions on CT are characteristic
- Early treatment critical; delayed treatment results in irreversible fibrosis and CKD
- High-dose IV steroids for severe AKI; many achieve partial (not complete) renal recovery
Case 4: Retroperitoneal Mass Suspicious for Malignancy
Presentation: A 58-year-old man presents with 2 months of lower back pain and left flank pain. CT abdomen performed for suspected renal colic shows large retroperitoneal soft tissue mass encasing the aorta and left ureter with hydronephrosis. Referred to oncology for suspected retroperitoneal sarcoma.
Examination: Left flank tenderness, no palpable masses, no lymphadenopathy, no stigmata of malignancy.
Initial Investigations:
- Normal inflammatory markers (ESR 18 mm/hr, CRP 6 mg/L)
- Creatinine 145 μmol/L (mildly elevated)
- CT abdomen: 8 cm periaortic soft tissue mass encasing abdominal aorta, medial deviation of ureters, left hydronephrosis
Differential Diagnosis:
- Retroperitoneal sarcoma
- Lymphoma
- IgG4-related retroperitoneal fibrosis
- Idiopathic retroperitoneal fibrosis
Key Investigations:
- Serum IgG4: 95 mg/dL (normal—does NOT exclude IgG4-RD)
- PET-CT: FDG-avid periaortic soft tissue (SUV max 6.2), mild uptake in pancreas, no distant lesions
- CT-guided core biopsy of retroperitoneal mass: Dense lymphoplasmacytic infiltrate, storiform fibrosis, obliterative phlebitis, IgG4+ plasma cells 90/HPF, IgG4/IgG ratio 55%
- Whole-body imaging: Diffuse mild pancreatic enlargement (asymptomatic type 1 AIP)
Diagnosis: IgG4-related retroperitoneal fibrosis with concurrent autoimmune pancreatitis
Management:
- Urgent ureteric stent placement for left hydronephrosis
- Prednisone 40 mg daily initiated
- Dramatic reduction in retroperitoneal mass size on CT at 3 months (8 cm → 4 cm)
- Resolution of hydronephrosis, stent removed at 6 months
- Steroid taper to 5 mg daily maintenance
- No relapse at 18 months
Learning Points:
- IgG4-RD can mimic malignancy (sarcoma, lymphoma); biopsy essential before treatment
- Normal serum IgG4 does NOT exclude IgG4-RD (30-40% have normal levels)
- Histology showing storiform fibrosis + obliterative phlebitis + IgG4+ cells > 40% is diagnostic
- Retroperitoneal fibrosis may cause ureteric obstruction requiring stent before medical therapy
Case 5: Steroid-Refractory Disease
Presentation: A 64-year-old woman with biopsy-proven IgG4-related sialadenitis and sclerosing cholangitis started on prednisone 40 mg daily. After 4 weeks, minimal improvement in gland size and persistent cholestatic liver enzymes.
Investigations:
- Baseline: IgG4 350 mg/dL, bilirubin 60 μmol/L, ALP 320 U/L
- Week 4: IgG4 340 mg/dL (minimal decrease), bilirubin 55 μmol/L, ALP 300 U/L (minimal improvement)
- Repeat CT: Minimal change in salivary gland size, persistent biliary strictures
Assessment: Steroid-refractory IgG4-RD (lack of expected dramatic response)
Differential Considerations:
- True steroid-refractory disease (rare)
- Incorrect diagnosis (consider re-biopsy to exclude malignancy)
- Established fibrosis (limited reversibility)
- Non-compliance (verify medication adherence)
Management:
- Re-biopsy: Confirmed IgG4-RD, no evidence of malignancy or alternative diagnosis
- Rituximab 1g IV on days 1 and 15 added to prednisone
- Response at 8 weeks: IgG4 decreased to 120 mg/dL, bilirubin 30 μmol/L, ALP 180 U/L
- Response at 6 months: Near-complete resolution of salivary gland enlargement, biliary strictures improved on MRCP
- Rituximab maintenance: Every 6 months
- Prednisone tapered to 5 mg daily, then discontinued
- Sustained remission at 24 months
Learning Points:
- Lack of steroid response should prompt reconsideration of diagnosis (exclude malignancy)
- True steroid-refractory IgG4-RD is rare; rituximab highly effective in this setting
- Rituximab produces slower but sustained responses compared to steroids
- Combination rituximab + steroids may be more effective than either alone for refractory disease
16. Imaging Atlas and Interpretation Guide
Pancreas (Type 1 Autoimmune Pancreatitis)
CT Findings:
Diffuse Type 1 AIP:
- Pancreatic enlargement: "Sausage-shaped" pancreas with smooth, diffuse enlargement
- Enhancement pattern: Delayed enhancement on arterial phase (hypodense initially, enhances on delayed phase)
- Capsule-like rim: Low-attenuation peripheral rim representing fibrous pseudocapsule (pathognomonic)
- Pancreatic duct: Diffusely narrowed "featureless" duct without marked dilation
- Peripancreatic fat: Preserved (no significant stranding unlike acute pancreatitis)
Focal Type 1 AIP:
- Mass-like focal pancreatic enlargement (often head)
- Mimics pancreatic adenocarcinoma
- Delayed enhancement, capsule-like rim help distinguish from cancer
- Biopsy essential if imaging atypical
MRI/MRCP Findings:
- T1-weighted: Hypointense (reduced signal from inflammation/fibrosis)
- T2-weighted: Hypointense peripheral rim (fibrous capsule)
- Diffusion-weighted imaging (DWI): Restricted diffusion in active inflammation (high signal on DWI, low on ADC)
- Post-gadolinium: Delayed homogeneous enhancement
- MRCP: Pancreatic duct narrowing, skip lesions rare
EUS Findings:
- Diffuse hypoechoic pancreatic parenchyma
- Hyperechoic foci or strands (fibrosis)
- Pancreatic duct narrowing without significant dilation
- Peripancreatic hypoechoic rim (correlates with capsule)
Key Differentiating Features from Pancreatic Cancer:
| Feature | Type 1 AIP | Pancreatic Cancer |
|---|---|---|
| Morphology | Diffuse "sausage-shaped" or focal symmetric | Focal irregular mass |
| Enhancement | Delayed homogeneous | Hypoenhancing throughout |
| Capsule-like rim | Present (low-attenuation rim) | Absent |
| Duct dilation | Minimal; diffusely narrowed | Marked upstream dilation |
| Vascular involvement | Rare; vessels encased but patent | Common; vessel occlusion/encasement |
| Serum IgG4 | Elevated (> 280 mg/dL in 40%) | Normal or mildly elevated |
| Response to steroids | Dramatic (if empiric trial done—NOT recommended) | None |
Biliary Tree (IgG4 Sclerosing Cholangitis)
MRCP/ERCP Findings:
Stricture Patterns:
- Type 1: Distal common bile duct stricture only (most common; concurrent with type 1 AIP)
- Type 2: Intrahepatic and extrahepatic biliary strictures (mimics PSC)
- Type 3: Hilar stricture (mimics cholangiocarcinoma)
- Type 4: Intrahepatic strictures only
Characteristics of Strictures:
- Long, smooth strictures: (differs from PSC's short, multifocal "beaded" strictures)
- Wall thickening: Circumferential, homogeneous bile duct wall thickening on CT/MRI
- Upstream dilation: Intrahepatic duct dilation proximal to stricture
- No filling defects: Unlike cholangiocarcinoma with intraluminal tumor
Key Differentiating Features from PSC and Cholangiocarcinoma:
| Feature | IgG4-SC | PSC | Cholangiocarcinoma |
|---|---|---|---|
| Stricture length | Long, smooth | Short, multifocal | Variable; mass-forming |
| Beading | Absent | Present ("beaded" appearance) | Absent |
| Wall thickening | Marked, circumferential | Minimal | Irregular, asymmetric |
| IBD association | No | Strong (70% have IBD) | No |
| p-ANCA | Negative | Positive (70-80%) | Negative |
| Serum IgG4 | Elevated | Normal | Normal or mildly elevated |
| CA19-9 | Elevated (both benign and malignant) | Mildly elevated | Markedly elevated (> 1000) |
| Concurrent AIP | 30-60% | No | No |
| Response to steroids | Yes | No | No |
Retroperitoneum and Aorta
CT Findings:
Retroperitoneal Fibrosis:
- "Mantle sign": Soft tissue encasing abdominal aorta, forming a circumferential "mantle" or "halo"
- Medial ureteral deviation: Ureters pulled medially (normal ureters lateral to psoas muscles)
- Hydronephrosis: Bilateral or unilateral from ureteric compression
- Psoas muscle involvement: Soft tissue infiltration, enlargement
- Enhancement: Homogeneous soft tissue enhancement
IgG4-Related Aortitis/Periaortitis:
- Aortic wall thickening: Circumferential or eccentric wall thickening (> 3 mm)
- Periaortic soft tissue: "Halo" of soft tissue density surrounding aorta
- Inflammatory aneurysm: Aneurysmal dilation + wall thickening
- Involvement sites: Most common in abdominal aorta; can involve thoracic aorta, arch vessels
MRI Findings:
- T1-weighted: Iso- to hypointense soft tissue
- T2-weighted: Hyperintense (active inflammation) or hypointense (fibrosis)
- Post-gadolinium: Enhancement of periaortic tissue and aortic wall
- DWI: Restricted diffusion in active inflammation
PET-CT Findings:
- FDG-avid: Intense uptake (SUV max > 2.5) in active inflammation
- Distribution: Periaortic soft tissue, aortic wall, retroperitoneal mass
- Monitoring response: Metabolic activity decreases with treatment
Key Differentiating Features from Other Causes:
| Feature | IgG4-RD | Atherosclerotic Aneurysm | Takayasu Arteritis |
|---|---|---|---|
| Patient age | 50-70 years | > 60 years | less than 40 years |
| Soft tissue "halo" | Present | Absent | Absent |
| Aortic wall thickening | Circumferential, smooth | Calcified, atheroma | Irregular, stenotic |
| Multi-organ involvement | Common (pancreas, kidneys, glands) | No | No (but other vessels) |
| Inflammatory markers | Normal/mildly elevated | Normal | Markedly elevated (ESR > 50) |
| Serum IgG4 | Elevated | Normal | Normal |
Kidneys (IgG4-Related Tubulointerstitial Nephritis)
CT Findings:
IgG4-TIN Patterns:
- Bilateral cortical lesions: Multiple low-attenuation wedge-shaped or round lesions in renal cortex
- Diffuse enlargement: Bilateral enlarged kidneys with diffuse hypoattenuation
- Focal mass: Solitary mass-like lesion (mimics renal cell carcinoma)
- Pelvic/ureteric involvement: Thickening of renal pelvis, ureter
MRI Findings:
- T1-weighted: Hypointense cortical lesions
- T2-weighted: Hyperintense lesions (inflammation)
- DWI: Restricted diffusion in active inflammation
- Post-gadolinium: Minimal or no enhancement (hypovascular compared to RCC)
Key Differentiating Features from Renal Cell Carcinoma:
| Feature | IgG4-TIN | Renal Cell Carcinoma |
|---|---|---|
| Morphology | Bilateral cortical lesions or diffuse | Solitary mass, unilateral |
| Enhancement | Minimal/none | Avid enhancement (hypervascular) |
| Attenuation | Low-attenuation (10-20 HU) | Variable (soft tissue density) |
| Multi-organ involvement | Common | No |
| Serum IgG4 | Elevated | Normal |
| Renal function | Impaired (elevated creatinine) | Usually preserved unless advanced |
Salivary and Lacrimal Glands (Mikulicz Disease)
CT/MRI Findings:
Salivary Glands:
- Bilateral symmetric enlargement: Parotid and submandibular glands (80% bilateral)
- Homogeneous enhancement: Uniform enhancement without focal lesions
- Preserved architecture: Glandular architecture maintained (vs destructive lymphoma)
- No calcifications: Unlike chronic sialadenitis (Sjögren often has punctate calcifications)
Lacrimal Glands:
- Bilateral enlargement: Symmetric involvement of lacrimal glands
- Homogeneous appearance: Uniform soft tissue density/signal
- No bony erosion: Orbital bone intact (vs aggressive tumor)
MRI Characteristics:
- T1-weighted: Iso- to hypointense
- T2-weighted: Hyperintense (inflammatory edema) or hypointense (fibrosis)
- Post-gadolinium: Homogeneous enhancement
- Diffusion: Restricted diffusion in active inflammation
Key Differentiating Features from Sjögren and Lymphoma:
| Feature | IgG4-RD (Mikulicz) | Sjögren Syndrome | Lymphoma (MALT) |
|---|---|---|---|
| Gland size | Markedly enlarged | Mildly to moderately enlarged | Enlarged, may be asymmetric |
| Enhancement | Homogeneous | Heterogeneous | Heterogeneous, infiltrative |
| Architecture | Preserved | May show cystic changes | Destructive, loss of architecture |
| Calcifications | Absent | May be present | Absent |
| IgG4+ cells | > 40% | less than 10% | Variable |
| Anti-SSA/SSB | Negative | Positive (60%) | Negative |
Meninges (IgG4-Related Pachymeningitis)
MRI Findings:
Pattern:
- Pachymeningeal thickening: Thickening of dura mater (outer meningeal layer)
- Enhancement: Strong, homogeneous enhancement with gadolinium
- Distribution: May be focal (mass-like) or diffuse
- Common sites: Frontal, parietal convexities; tentorium; skull base
T1/T2 Characteristics:
- T1-weighted: Iso- to hypointense thickened dura
- T2-weighted: Iso- to hyperintense
- FLAIR: Hyperintense
Key Differentiating Features:
| Feature | IgG4-RD Pachymeningitis | GPA (Wegener's) | Neurosarcoidosis | Meningioma |
|---|---|---|---|---|
| Pattern | Pachymeningeal (dural) | Pachymeningeal | Leptomeningeal > pachymeningeal | Mass, extra-axial |
| Distribution | Focal or diffuse | Often skull base | Basilar, nodular | Solitary mass |
| Enhancement | Homogeneous | Homogeneous | Nodular | Intense, homogeneous |
| Multi-organ involvement | Common | Yes (lung, kidney, sinuses) | Yes (lungs, lymph nodes) | No |
| Serum IgG4 | Elevated | Normal | Normal | Normal |
| ANCA | Negative | c-ANCA/PR3 positive | Negative | Negative |
17. Multidisciplinary Team (MDT) Management
IgG4-RD management requires collaboration across multiple specialties due to multi-organ involvement.
Core MDT Members
Rheumatology:
- Lead coordinator for systemic immunosuppression
- Initiation and monitoring of glucocorticoids, rituximab, steroid-sparing agents
- Long-term follow-up and relapse management
- Bone protection, management of steroid side effects
Gastroenterology/Hepatology:
- Management of autoimmune pancreatitis type 1
- ERCP with biliary stenting for IgG4 sclerosing cholangitis
- Pancreatic enzyme replacement for exocrine insufficiency
- Endoscopic ultrasound with biopsy
Nephrology:
- Management of IgG4-tubulointerstitial nephritis
- Renal biopsy for diagnosis
- Management of acute kidney injury, chronic kidney disease
- Ureteric stent placement (with urology)
Radiology:
- Cross-sectional imaging (CT, MRI, MRCP)
- PET-CT for multi-organ assessment and treatment response
- Image-guided biopsy
- Interpretation and differential diagnosis (IgG4-RD vs malignancy)
Histopathology:
- Tissue diagnosis with immunohistochemistry (IgG4, IgG staining)
- Differentiation from malignancy, other inflammatory conditions
- Quantification of IgG4+ cells and IgG4/IgG ratio
Vascular Surgery:
- Management of aortic aneurysms, inflammatory aneurysms
- Surgical intervention if aneurysm > 5.5 cm or symptomatic
- Preoperative steroid optimization
Ophthalmology:
- Assessment and monitoring of orbital disease, lacrimal involvement
- Visual acuity testing, optic nerve examination
- Urgent assessment if vision-threatening disease
Otolaryngology (ENT):
- Salivary gland biopsy (parotid, submandibular)
- Assessment of head and neck involvement
- Management of airway compromise from thyroid/laryngeal involvement
Urology:
- Ureteric stent placement for hydronephrosis
- Management of retroperitoneal fibrosis-related urinary obstruction
Neurology/Neurosurgery:
- Assessment of pachymeningitis, hypophysitis
- Management of neurological deficits
- Surgical decompression if indicated
MDT Meeting Structure
Discussion Points for Each Case:
-
Clinical Presentation and Diagnosis:
- Presenting symptoms, organ involvement
- Imaging findings (CT, MRI, PET-CT)
- Biopsy results, IgG4 immunostaining
- Serum IgG4 level
- Exclusion of malignancy and alternative diagnoses
-
Disease Activity and Severity:
- Active (proliferative) vs established (fibrotic) disease
- Single-organ vs multi-organ involvement
- Organ dysfunction severity (renal failure, biliary obstruction, vision loss)
- IgG4-RD Responder Index score (if calculated)
-
Treatment Plan:
- Need for urgent interventions (ERCP, ureteric stent, vascular surgery)
- Induction therapy (glucocorticoids ± rituximab)
- Maintenance strategy (steroid taper, rituximab, steroid-sparing agents)
- Prophylaxis (PJP, bone protection, vaccinations)
-
Monitoring Plan:
- Clinical follow-up schedule
- Biochemical monitoring (IgG4, organ-specific markers)
- Imaging intervals (CT/MRI/PET-CT)
- Surveillance for relapse, side effects
-
Relapse Management:
- Recognition of relapse (clinical, biochemical, radiological)
- Re-induction strategy (glucocorticoids vs rituximab)
- Escalation to combination therapy or alternative agents
18. Quality Improvement and Audit Standards
Key Performance Indicators (KPIs) for IgG4-RD Management
Diagnostic KPIs:
- Time to diagnosis: From symptom onset to confirmed diagnosis (less than 6 months target)
- Tissue diagnosis rate: Percentage of patients with histological confirmation (target > 90%)
- IgG4 immunostaining performed: In all biopsies where IgG4-RD suspected (target 100%)
- Malignancy excluded: Documented exclusion of malignancy before immunosuppression (target 100%)
- Multi-organ screening: CT chest/abdomen/pelvis performed at baseline (target > 90%)
Treatment KPIs:
- Steroid induction within 2 weeks: Of confirmed diagnosis (target > 90%)
- Treatment response assessment: Clinical/biochemical/radiological assessment at 3-6 months (target 100%)
- Rituximab for relapsing disease: Offered to patients with relapse or high-risk features (target > 80%)
- Bone protection: Calcium/vitamin D + bisphosphonates for patients on steroids > 3 months (target > 90%)
- PJP prophylaxis: For patients on steroids > 20 mg/day for > 1 month (target 100%)
Follow-Up KPIs:
- Relapse detection: Clinical/biochemical monitoring every 3-6 months (target > 90%)
- Imaging surveillance: Repeat imaging at 6-12 months intervals (target > 80%)
- Long-term follow-up: Annual review for at least 5 years (target > 80%)
- Adverse event monitoring: Documentation of steroid/rituximab side effects (target 100%)
Audit Questions
- What percentage of patients with suspected IgG4-RD undergo tissue biopsy before starting immunosuppression?
- What is the median time from presentation to diagnosis?
- What proportion of patients achieve remission with first-line glucocorticoid therapy?
- What is the relapse rate at 1 year, 2 years, 5 years?
- What percentage of patients with relapsing disease receive rituximab?
- What proportion of patients on long-term steroids receive bone protection?
- What is the rate of serious adverse events (infection, malignancy, death)?
19. Research Frontiers and Future Directions
Current Research Areas
Biomarker Development:
- Circulating IgG4+ plasmablasts as predictors of disease activity and relapse
- Novel autoantibodies (anti-galectin-3, anti-laminin 511-E8, anti-prohibitin) for diagnosis
- Cytokine profiles (IL-4, IL-10, IL-13, TGF-β) as disease activity markers
- Metabolomic and proteomic signatures
Pathogenesis Studies:
- Identification of initiating antigens and environmental triggers
- Role of specific T cell subsets (Tfh2 vs Tfh1, CD4 CTLs, Tregs) in different phenotypes
- Mechanisms of IgG4 antibody pathogenicity (blocking vs immune complex formation)
- Fibrosis pathways and potential reversibility
Therapeutic Trials:
- B cell-targeting: Comparison of rituximab regimens (dose, frequency, maintenance strategies)
- T cell-targeting: Abatacept (CTLA-4-Ig), other T cell co-stimulation blockers
- Cytokine inhibition: Anti-IL-4, anti-IL-13, anti-IL-6 trials
- Small molecules: JAK inhibitors, BTK inhibitors targeting B cell signaling
- Plasma cell-targeting: Bortezomib, newer proteasome inhibitors
Genetic and Genomic Studies:
- Genome-wide association studies (GWAS) to identify susceptibility loci
- Transcriptomic profiling of affected tissues
- Epigenetic modifications in IgG4-RD
- HLA typing across different populations and phenotypes
Long-Term Outcomes Research:
- Cancer risk in IgG4-RD: causation vs association
- Cardiovascular outcomes in patients with aortitis
- Quality of life and patient-reported outcome measures
- Optimal monitoring strategies for relapse detection
Unanswered Questions
- What initiates IgG4-RD? Environmental triggers, infectious agents, or purely genetic susceptibility?
- Why IgG4? What is the specific role of IgG4 antibodies in pathogenesis given their typically non-inflammatory properties?
- Can fibrosis be reversed? What is the window for treatment to prevent irreversible organ damage?
- Can we predict relapse? Which patients can safely discontinue therapy vs require indefinite maintenance?
- Optimal treatment duration? How long to continue maintenance rituximab or low-dose steroids?
- Malignancy relationship? Is increased cancer risk real, and if so, is it disease- or treatment-related?
20. Summary and Key Messages
Top 10 Take-Home Points
-
IgG4-RD is a multi-organ fibroinflammatory condition characterised by tumefactive lesions, IgG4+ plasma cell infiltration, storiform fibrosis, and obliterative phlebitis—recognized as a unified disease entity since 2003.
-
Diagnosis requires histopathology: Tissue biopsy showing dense lymphoplasmacytic infiltrate, storiform fibrosis, obliterative phlebitis, and IgG4+ plasma cells > 40% of IgG+ cells is essential for definitive diagnosis.
-
Serum IgG4 alone is insufficient: Elevated in only 60-70% of cases; normal levels do NOT exclude IgG4-RD. IgG4 > 280 mg/dL is highly suggestive but requires clinical and histological correlation.
-
IgG4-RD mimics malignancy: Pancreatic, biliary, and retroperitoneal manifestations closely resemble cancer. Always exclude malignancy with tissue diagnosis before initiating immunosuppression.
-
First-line treatment is glucocorticoids: Prednisone 40 mg/day (0.6 mg/kg/day) produces dramatic responses in > 90% of patients, often within days to weeks. Lack of response should prompt diagnostic reconsideration.
-
Relapse is common: 30-60% relapse after glucocorticoid withdrawal, particularly with rapid taper or multi-organ disease. Rituximab maintenance reduces relapse to 20-30%.
-
Rituximab is highly effective second-line therapy: For relapsing disease, steroid-refractory cases, and maintenance. Superior to conventional immunosuppressants for relapse prevention.
-
Early treatment prevents irreversible fibrosis: Proliferative (inflammatory) disease is reversible with treatment; established fibrosis has limited reversibility. Prompt diagnosis and treatment are critical.
-
Multi-organ screening is essential: 60-90% have multi-organ involvement at diagnosis or develop it over time. Perform whole-body imaging (CT chest/abdomen/pelvis) at baseline.
-
Multidisciplinary management is key: IgG4-RD crosses traditional specialty boundaries. Rheumatology coordinates systemic therapy, but gastroenterology, nephrology, radiology, pathology, and other specialties are essential for optimal care.
Last Updated: 2026-01-16 Evidence Level: High (33 PubMed-indexed citations) Target Examinations: MRCP, FRACP, MRCPCH, USMLE Step 2/3, PLAB Difficulty: Moderate to High Estimated Reading Time: 45-60 minutes Line Count: 1,784 lines