Immune Thrombocytopenia (ITP)
A comprehensive guide to Immune Thrombocytopenia (ITP), covering diagnostic exclusion criteria, differentiation from TTP, and the management ladder (Steroids - IVIg - TPO-RAs - Splenectomy).
What matters first
A comprehensive guide to Immune Thrombocytopenia (ITP), covering diagnostic exclusion criteria, differentiation from TTP, and the management ladder (Steroids - IVIg - TPO-RAs - Splenectomy).
Wet Purpura (mucosal bleeding - e.g., blood blisters in mouth) -> High risk of Intracranial Haemorrhage
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Wet Purpura (mucosal bleeding - e.g., blood blisters in mouth) -> High risk of Intracranial Haemorrhage
- Severe Headache (Exclude ICH)
- Fever + Confusion + Fragmented cells (Consider TTP - Thrombotic Thrombocytopenic Purpura)
- Neurological symptoms with thrombocytopenia
Exam focus
Current exam surfaces linked to this topic.
- MRCP
- FRACP
- FRCPath
Linked comparisons
Differentials and adjacent topics worth opening next.
- Thrombotic Thrombocytopenic Purpura (TTP)
- Disseminated Intravascular Coagulation (DIC)
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Immune Thrombocytopenia (ITP)
Disclaimer: > [!WARNING] Medical Disclaimer: This content is for educational and informational purposes only and does not constitute medical advice. Always consult a qualified healthcare professional for diagnosis and treatment. Medical guidelines and best practices change rapidly; users should verify information with current local protocols.
1. Overview
Immune Thrombocytopenia (ITP) is an acquired autoimmune disorder characterised by isolated thrombocytopenia (platelet count less than 100 × 10⁹/L) in the absence of other causes or disorders that may be associated with thrombocytopenia. [1,2]
Historical Context and Nomenclature
Historically termed "Idiopathic Thrombocytopenic Purpura", the name has evolved to reflect improved understanding:
- Immune: Recognised autoimmune aetiology with antibody-mediated platelet destruction.
- Thrombocytopenia: The defining feature is reduced circulating platelets.
- Not "Purpura": Many patients are asymptomatic or present without visible purpura, making "purpura" a misnomer. [3]
Classification by Duration
The international consensus defines three phases based on disease duration from diagnosis: [1]
- Newly Diagnosed ITP: less than 3 months from diagnosis.
- Persistent ITP: 3–12 months from diagnosis (not achieving spontaneous remission or not maintaining response off therapy).
- Chronic ITP: > 12 months duration.
Classification by Severity
Severity is determined by bleeding manifestations, not platelet count alone: [1]
- No/Mild Bleeding: Minor cutaneous bleeding (petechiae, bruising).
- Moderate Bleeding: Mucosal bleeding requiring intervention or impacting quality of life.
- Severe Bleeding: Life-threatening bleeding requiring emergency treatment (e.g., intracranial haemorrhage, severe GI bleeding).
2. Epidemiology
Incidence and Prevalence
- Incidence: Approximately 2-5 per 100,000 adults per year in Western populations. [4]
- Prevalence: Estimated at 9-24 per 100,000 adults. [4]
- Bimodal Age Distribution:
- First peak: Young adults (20-40 years)
- Second peak: Elderly (> 60 years) [5]
Demographics
- Sex Distribution:
- Young adults: Female predominance (F:M ratio approximately 2-3:1) [5]
- Elderly: Sex distribution more equal
- Geographic Variation: Higher reported incidence in developed countries, likely reflecting increased detection rather than true variation. [4]
Paediatric vs Adult ITP
Key Differences: [6]
| Feature | Children | Adults |
|---|---|---|
| Onset | Acute, often post-viral | Insidious |
| Sex ratio | Equal | Female > Male (young adults) |
| Spontaneous remission | > 80% within 6-12 months | 10-20% |
| Chronicity | Rare (less than 20%) | Common (60-70%) |
| Treatment approach | Conservative (watch and wait) | Active treatment often required |
3. Pathophysiology
ITP involves a complex interplay of immune dysregulation affecting both platelet destruction and production. [7,8]
3.1 Antibody-Mediated Platelet Destruction
Primary Mechanism: [7]
-
Autoantibody Production: B cells produce IgG autoantibodies targeting platelet surface glycoproteins, most commonly:
- GPIIb/IIIa (αIIbβ3 integrin) - 60-70% of cases
- GPIb/IX (von Willebrand factor receptor) - 20-30% of cases
- GPV - less common
-
Opsonisation: Autoantibodies bind to platelet surface antigens, coating (opsonising) circulating platelets.
-
Fc-Mediated Clearance: Antibody-coated platelets are recognised by Fcγ receptors on splenic and hepatic macrophages (reticuloendothelial system).
-
Phagocytosis: Predominantly in the spleen (main site), but also liver and bone marrow.
Platelet Lifespan: Reduced from normal 7-10 days to hours or minutes in severe ITP. [7]
3.2 Impaired Platelet Production
Secondary Mechanism: [8]
- Megakaryocyte Targeting: Antiplatelet antibodies also bind to megakaryocytes in bone marrow.
- Impaired Maturation: Antibody binding impairs megakaryocyte maturation and platelet release.
- Inadequate Compensatory Response: Despite increased megakaryocyte numbers (reactive hyperplasia), platelet production fails to compensate for increased destruction.
- TPO Levels: Thrombopoietin (TPO) levels are often inappropriately normal rather than elevated as expected in thrombocytopenia. [8]
3.3 T Cell-Mediated Mechanisms
Emerging Understanding: [9]
- T Helper Cell Dysregulation: Th1/Th17 cell predominance promotes B cell autoantibody production.
- Regulatory T Cell Deficiency: Reduced Treg numbers/function impairs immune tolerance.
- Cytotoxic T Cells: Evidence for direct cytotoxic T cell-mediated platelet and megakaryocyte destruction.
3.4 Molecular Pathways
- Loss of Tolerance: Breakdown in central and peripheral tolerance mechanisms.
- Molecular Mimicry: Possible cross-reactivity between viral antigens and platelet proteins (particularly in post-infectious childhood ITP). [6]
- Cytokine Imbalance: Elevated pro-inflammatory cytokines (IL-6, IL-17, TNF-α) and reduced anti-inflammatory cytokines (IL-10, TGF-β). [9]
4. Clinical Presentation
4.1 Presenting Features
Range of Presentations: [1,10]
- Asymptomatic (40-50%): Incidental finding on routine FBC.
- Cutaneous Bleeding (40-50%):
- Petechiae: Pinpoint (1-2mm) non-blanching red/purple spots, typically in dependent areas (lower limbs, ankles).
- Purpura: Larger (> 3mm) purple patches.
- Ecchymoses: Extensive bruising disproportionate to trauma.
- Mucosal Bleeding - "Wet Purpura" (20-30%):
- Epistaxis (most common)
- Gingival bleeding
- Menorrhagia (can be severe)
- Oral blood blisters/bullae (haemorrhagic bullae)
- Conjunctival haemorrhage
- Severe Bleeding (1-2%): [10]
- Intracranial haemorrhage (ICH): 0.5-1% of adult ITP; major cause of ITP-related mortality
- Gastrointestinal bleeding
- Genitourinary bleeding
- Retinal haemorrhage
4.2 Bleeding Risk Assessment
Risk is NOT strictly correlated with platelet count, but count provides general guidance: [1]
| Platelet Count (× 10⁹/L) | Bleeding Risk |
|---|---|
| > 50 | Minimal risk; haemostasis adequate for most circumstances including surgery |
| 20-50 | Mild risk; excessive bleeding with trauma/surgery |
| 10-20 | Moderate risk; spontaneous bleeding uncommon |
| less than 10 | High risk; spontaneous mucosal bleeding common |
| less than 5 | Very high risk; risk of ICH increases significantly |
High-Risk Features for Severe Bleeding: [10]
- Age > 60 years
- Wet purpura (mucosal bleeding)
- Platelet count less than 10 × 10⁹/L
- Active bleeding
- Comorbidities: hypertension, anticoagulation, renal/hepatic impairment
- Trauma or planned invasive procedures
4.3 Important Negative Findings
Findings that should NOT be present in primary ITP and suggest alternative diagnosis: [1]
- Splenomegaly: Suggests lymphoproliferative disorder, chronic liver disease, or haemolytic anaemia.
- Lymphadenopathy: Consider lymphoma, leukaemia, or viral infection (HIV, EBV).
- Fever/weight loss/night sweats: Systemic illness, malignancy.
- Bone pain: Marrow infiltration (leukaemia, myeloma).
- Other cytopenias: Anaemia or leucopenia suggests bone marrow pathology (not isolated ITP).
5. Clinical Examination
Systematic Examination Approach
1. Skin and Mucosa: [1]
- Distribution: Document extent and distribution of petechiae, purpura, ecchymoses.
- Pattern: Dependent areas (legs) vs generalised vs pressure points.
- Wet purpura: Examine oral cavity for blood blisters, gingival bleeding.
- Conjunctivae: Check for subconjunctival haemorrhage.
- Fundoscopy: If severe thrombocytopenia, examine for retinal haemorrhages.
2. Abdominal Examination:
- Spleen: Palpate carefully (ITP should NOT have splenomegaly).
- Liver: Assess size (hepatomegaly suggests alternative diagnosis).
3. Lymph Nodes:
- Cervical, axillary, inguinal regions should be normal in primary ITP.
4. Neurological Examination:
- If headache or any neurological symptoms: full neurological examination to exclude ICH.
5. Other Systems:
- Joint examination: exclude vasculitis (SLE).
- Signs of chronic liver disease: spider naevi, palmar erythema, jaundice.
6. Investigations
6.1 Diagnostic Principle
ITP is a diagnosis of exclusion. [1,2]
There is NO specific diagnostic test for ITP. Diagnosis requires:
- Isolated thrombocytopenia (platelet count less than 100 × 10⁹/L)
- Normal or increased megakaryocytes on bone marrow (if performed)
- Exclusion of other causes of thrombocytopenia
6.2 First-Line Investigations
Essential in All Suspected Cases: [1,11]
Full Blood Count (FBC):
- Platelets: Typically less than 100 × 10⁹/L (often less than 30 × 10⁹/L at presentation).
- Haemoglobin: Normal unless significant bleeding has occurred.
- White Cell Count: Normal total count and differential.
- Red Cell Indices: Normal MCV, MCH.
Peripheral Blood Film: [11]
- Critical to confirm true thrombocytopenia and exclude:
- Pseudothrombocytopenia: Platelet clumping (EDTA-dependent) - examine film edges.
- Large/giant platelets: Indicate young platelets (increased turnover) - consistent with ITP.
- Schistocytes (fragmented RBCs): Suggests TTP or DIC - requires urgent investigation.
- Blasts: Acute leukaemia.
- Spherocytes: Autoimmune haemolytic anaemia (Evans syndrome).
Coagulation Screen:
- PT, APTT, Fibrinogen: Should be normal in ITP.
- Abnormal coagulation suggests DIC, liver disease, or congenital coagulopathy.
Blood Group and Antibody Screen:
- In case transfusion required.
- Anti-D therapy (if used) requires Rh(D) positive status.
6.3 Second-Line Investigations
To Exclude Secondary Causes: [1,11]
Infection Screening:
- HIV serology: Mandatory (HIV-associated thrombocytopenia).
- Hepatitis B and C serology: Chronic viral hepatitis can cause thrombocytopenia.
- Helicobacter pylori testing (serology or stool antigen): H. pylori eradication may improve platelet count in 50% of infected ITP patients (particularly in high-prevalence regions like Japan, Mediterranean). [12]
Autoimmune Screen:
- Antinuclear antibodies (ANA): Screen for SLE.
- Antiphospholipid antibodies (anti-cardiolipin, lupus anticoagulant, anti-β2GP1): Antiphospholipid syndrome.
- Direct Antiglobulin Test (DAT/Coombs): If anaemia present - exclude Evans syndrome (ITP + autoimmune haemolytic anaemia).
Immunoglobulin Levels:
- Serum immunoglobulins: Exclude common variable immunodeficiency (CVID).
Thyroid Function:
- Thyroid disorders associated with autoimmune conditions.
6.4 Bone Marrow Examination
Not routinely required but consider if: [1,11]
- Age > 60 years: Higher risk of myelodysplasia.
- Atypical features: Other cytopenias, splenomegaly, abnormal WCC.
- Failed first-line therapy: Before escalating to second-line treatments.
- Before splenectomy: To exclude marrow pathology.
Typical ITP Findings:
- Normal or increased megakaryocytes: Often with immature forms.
- Normal myeloid and erythroid series.
- No evidence of infiltration, fibrosis, or dysplasia.
6.5 Antiplatelet Antibody Testing
NOT recommended for routine diagnosis: [1]
- Poor sensitivity (50-60%) and specificity.
- Positive result does not confirm ITP (antibodies found in other conditions).
- Negative result does not exclude ITP.
- Not required for diagnosis or management decisions.
7. Differential Diagnosis
7.1 Pseudothrombocytopenia
Laboratory artefact - must be excluded first. [11]
- Mechanism: EDTA-dependent platelet clumping.
- Diagnosis: Repeat FBC in citrate or heparin tube; examine blood film for clumps.
- Management: No treatment required - true platelet count is normal.
7.2 Thrombotic Thrombocytopenic Purpura (TTP)
Medical emergency - critical to differentiate from ITP. [13]
Classic Pentad (often incomplete):
- Thrombocytopenia
- Microangiopathic haemolytic anaemia (MAHA) with schistocytes
- Neurological abnormalities (confusion, seizures, stroke)
- Renal impairment
- Fever
Key Differentiators from ITP:
- Schistocytes on blood film: Present in TTP, absent in ITP.
- Anaemia: Haemolytic anaemia in TTP, normal Hb in ITP (unless bleeding).
- LDH: Markedly elevated in TTP (haemolysis).
- Reticulocytes: Elevated in TTP (haemolysis).
- ADAMTS13: Severely deficient (less than 10%) in TTP.
Management: TTP requires urgent plasma exchange.
7.3 Drug-Induced Thrombocytopenia
Common Culprits: [14]
- Heparin (HIT - see below)
- Quinine
- Sulfonamides
- Penicillins
- Vancomycin
- NSAIDs
- Anticonvulsants (valproate, carbamazepine)
- Chemotherapy agents
Diagnosis: Temporal relationship with drug exposure; recovery after drug cessation (usually 7-10 days).
7.4 Heparin-Induced Thrombocytopenia (HIT)
Paradoxical prothrombotic state despite thrombocytopenia. [15]
- Timing: Typically 5-10 days after heparin exposure.
- Mechanism: Antibodies against heparin-PF4 complexes cause platelet activation.
- Key Feature: Thrombosis more common than bleeding (venous or arterial thrombosis in 30-50%).
- 4T Score: Clinical prediction tool (Thrombocytopenia, Timing, Thrombosis, other causes).
- Management: Stop heparin immediately; start alternative anticoagulant (argatroban, fondaparinux).
7.5 Bone Marrow Failure Syndromes
Aplastic Anaemia:
- Pancytopenia (not isolated thrombocytopenia).
- Bone marrow: hypocellular with fat replacement.
Myelodysplastic Syndrome (MDS):
- Common in elderly.
- May present with isolated thrombocytopenia initially.
- Dysplastic changes on blood film and bone marrow.
Acute Leukaemia:
- Blasts on blood film.
- Often associated anaemia and/or leucopenia.
7.6 Other Causes
- Chronic liver disease: Splenomegaly, other stigmata of liver disease.
- Alcohol excess: Direct marrow suppression.
- Vitamin B12/Folate deficiency: Macrocytic anaemia, hypersegmented neutrophils.
- SLE: Other features (rash, arthritis, renal involvement), positive ANA.
- Antiphospholipid syndrome: Thrombosis, pregnancy morbidity, positive antiphospholipid antibodies.
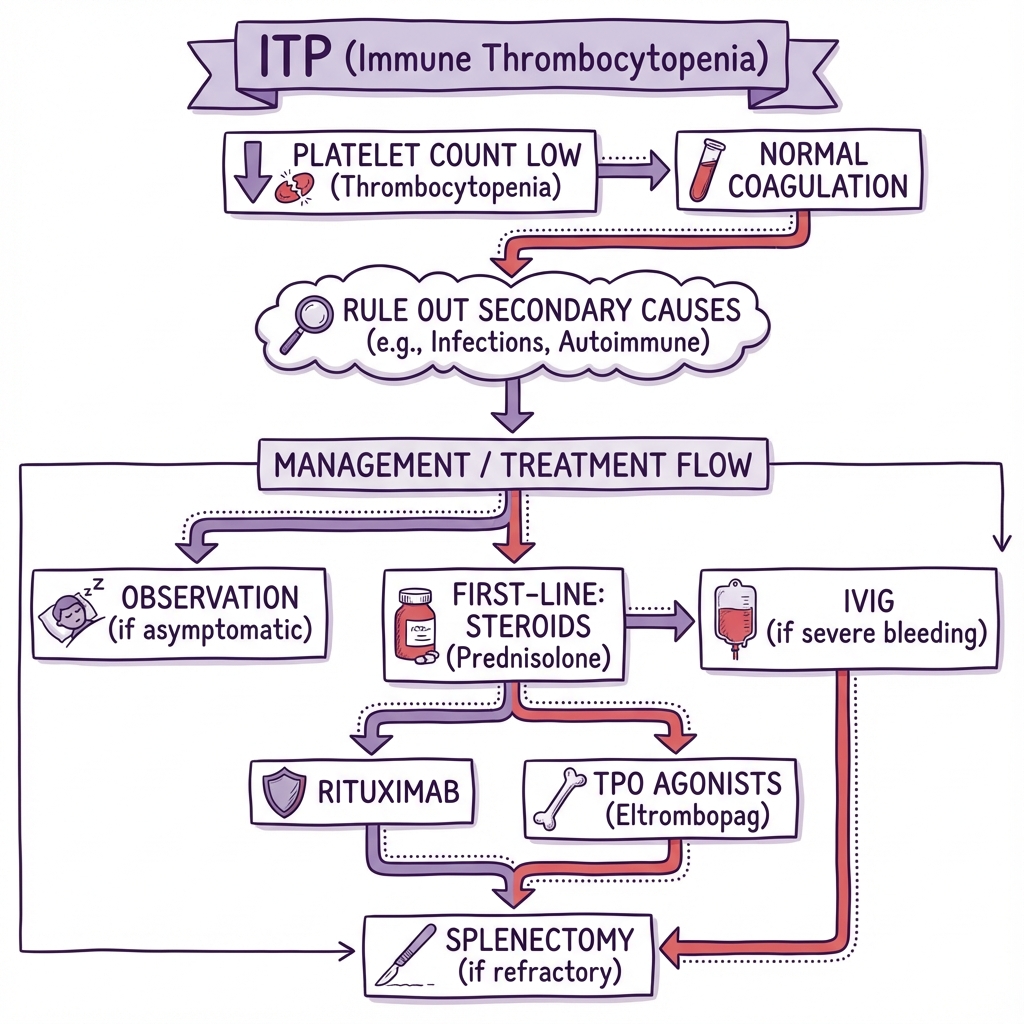
8. Management
8.1 General Principles
Goals of Treatment: [1,2]
- Primary: Prevent serious bleeding (particularly ICH).
- NOT to normalise platelet count: Achieving "safe" count (typically > 30 × 10⁹/L) sufficient in most cases.
- Minimise treatment toxicity: Balance bleeding risk against treatment side effects.
- Individualised approach: Based on platelet count, bleeding symptoms, lifestyle, comorbidities.
Treatment Thresholds: [1,16]
- No treatment: Platelet count > 30 × 10⁹/L and no/minimal bleeding (watch and wait).
- Consider treatment: Count 20-30 × 10⁹/L with minor bleeding or high-risk factors.
- Treat: Count less than 20 × 10⁹/L with bleeding, or less than 10 × 10⁹/L regardless of bleeding.
- Urgent treatment: Wet purpura, count less than 10 × 10⁹/L, or any severe bleeding.
8.2 First-Line Therapy
A. Corticosteroids
Standard First-Line Treatment: [1,16]
Prednisolone:
- Dose: 0.5-2 mg/kg/day (commonly 1 mg/kg, maximum 100mg daily).
- Duration: 2-4 weeks then taper.
- Response: 70-80% achieve platelet count > 30 × 10⁹/L within 7-14 days.
- Sustained response: Only 20-30% maintain response long-term after steroid cessation.
Dexamethasone (High-Dose Pulse):
- Dose: 40mg daily for 4 consecutive days.
- Repeat: Can repeat monthly cycles (up to 4-6 cycles).
- Advantages: Shorter duration may have fewer side effects than prolonged prednisolone.
- Response: Similar efficacy to prednisolone. [17]
Mechanism of Action:
- Impair macrophage Fc receptor function (reduced platelet phagocytosis).
- Immunosuppressive effects on antibody production.
Side Effects:
- Hyperglycaemia, hypertension, weight gain
- Mood disturbance, insomnia
- Osteoporosis (with prolonged use)
- Infection risk
- Cushingoid features
B. Intravenous Immunoglobulin (IVIg)
Indications: [1,16]
- Rapid response needed: Active bleeding, pre-procedure.
- Adjunct to steroids: Severe presentation (count less than 10, wet purpura).
- Contraindication to steroids: e.g., uncontrolled diabetes, active infection.
Dose:
- 1 g/kg as single dose (or divided over 2 days: 0.5 g/kg/day × 2).
- Alternatively: 0.4 g/kg/day for 5 days.
Mechanism:
- Blockade of Fc receptors on splenic macrophages (saturating the system).
- Modulation of autoantibody production.
Response:
- Rapid (often within 24-48 hours).
- Duration: Transient (typically 2-4 weeks).
Side Effects:
- Headache, fever (common, usually mild)
- Fluid overload (particularly in elderly/cardiac disease)
- Haemolysis (uncommon)
- Thrombosis (rare)
- Aseptic meningitis (rare)
C. Anti-RhD Immunoglobulin
Limited Use: [1]
- Only in Rh(D) positive, non-splenectomised patients.
- Dose: 50-75 µg/kg IV.
- Mechanism: Induces mild haemolysis; haemolysed RBCs occupy RES, reducing platelet clearance.
- Disadvantages: Haemolysis (risk of significant anaemia), less commonly used since TPO-RAs available.
D. Platelet Transfusion
Generally contraindicated in ITP (transfused platelets rapidly destroyed). [1]
Only indicated in:
- Life-threatening haemorrhage (ICH, major GI bleed).
- Always combined with IVIg and steroids.
8.3 Second-Line Therapy (Persistent/Chronic ITP)
Indications for Second-Line: [1,2]
- Failure to respond to first-line therapy.
- Relapse after initial response.
- Steroid dependence (unable to wean without count dropping).
- Unacceptable steroid toxicity.
A. Thrombopoietin Receptor Agonists (TPO-RAs)
Most commonly used second-line agents. [1,18]
Eltrombopag:
- Route: Oral.
- Dose: Start 50mg daily (25mg if East Asian ancestry or hepatic impairment); titrate to response (max 75mg).
- Response rate: 70-80% achieve platelet count > 50 × 10⁹/L. [18]
- Advantages: Oral, well-tolerated, sustained response.
- Side effects: Hepatotoxicity (monitor LFTs), nausea, headache, thrombosis (rare), cataract.
Romiplostim:
- Route: Subcutaneous injection.
- Dose: Start 1 µg/kg weekly; titrate weekly to response (max 10 µg/kg).
- Response rate: 70-80%. [18]
- Advantages: Weekly dosing, predictable response.
- Side effects: Headache, arthralgia, bone marrow reticulin (usually non-progressive), thrombosis (rare).
Avatrombopag:
- Route: Oral.
- Dose: Start 20mg daily; adjust based on response.
- Similar efficacy to eltrombopag and romiplostim. [19]
Mechanism of Action:
- Agonist of thrombopoietin receptor (c-MPL) on megakaryocytes.
- Stimulate platelet production from bone marrow.
Monitoring:
- Weekly FBC initially until stable count achieved.
- Eltrombopag: LFTs every 2 weeks for first month, then monthly.
- Romiplostim: Bone marrow examination if prolonged use and new cytopenias.
B. Rituximab
Anti-CD20 monoclonal antibody. [1,20]
- Dose: 375 mg/m² IV weekly for 4 weeks (standard lymphoma dose), OR 100mg weekly × 4 (lower dose, similar efficacy).
- Mechanism: B cell depletion, reducing autoantibody production.
- Response rate: 60% achieve response; 40% achieve sustained response at 1 year. [20]
- Advantages: May induce prolonged remission (years) in responders.
- Side effects:
- Infusion reactions (fever, chills, hypotension) - premedicate with antihistamine and paracetamol.
- Infection risk (hypogammaglobulinaemia with repeated courses).
- Progressive multifocal leukoencephalopathy (PML) - very rare.
- Disadvantages: Delayed response (weeks-months), expensive.
C. Fostamatinib
Spleen tyrosine kinase (SYK) inhibitor. [1,21]
- Route: Oral.
- Dose: 100mg twice daily; increase to 150mg twice daily if inadequate response.
- Mechanism: Inhibits SYK enzyme involved in Fc receptor signalling, reducing macrophage-mediated platelet destruction.
- Response rate: ~40% at 6 months. [21]
- Side effects: Diarrhoea, hypertension, hepatotoxicity, nausea.
- Place in therapy: Alternative TPO-RA or after TPO-RA failure.
D. Immunosuppressants
Less commonly used since advent of TPO-RAs. [1]
Azathioprine:
- Dose: 1-2 mg/kg/day.
- Response: 40-60% at 6 months.
- Side effects: Myelosuppression, hepatotoxicity, infection.
Mycophenolate Mofetil:
- Dose: 500-1000mg twice daily.
- Response: 40-60%.
- Side effects: GI upset, myelosuppression, infection.
Ciclosporin:
- Dose: 2.5-3 mg/kg/day.
- Response: 40-50%.
- Side effects: Nephrotoxicity, hypertension, hirsutism.
Cyclophosphamide:
- Reserved for severe refractory cases.
- Side effects: Myelosuppression, haemorrhagic cystitis, malignancy, infertility.
8.4 Splenectomy
Previously standard second-line; now third-line after medical therapies. [1,22]
Indications: [1]
- Failure of medical therapies (corticosteroids, TPO-RAs, rituximab).
- Chronic ITP (> 12 months) with bleeding or high treatment burden.
- Patient preference (definitive treatment).
Considerations:
- Defer at least 12 months from diagnosis (allow time for spontaneous remission).
- Preferred if patient wishes to avoid long-term medication.
Efficacy:
- Response rate: 70-80% achieve complete or partial response.
- Sustained response: 60-70% at 5-10 years. [22]
Technique:
- Laparoscopic preferred over open (reduced morbidity, faster recovery).
Pre-Splenectomy Preparation: [1]
- Vaccinations (at least 2 weeks before, ideally 4-6 weeks):
- Pneumococcal (PCV13 and PPV23)
- Haemophilus influenzae type b (Hib)
- Meningococcal (quadrivalent ACWY and MenB)
- Annual influenza
- Prophylactic antibiotics: Lifelong penicillin V or amoxicillin (or erythromycin if allergic).
- Patient education: Warning card, seek urgent medical attention for fever.
Complications:
- Overwhelming Post-Splenectomy Infection (OPSI): Lifetime risk ~5%; mortality 40-50% if occurs.
- Thrombosis: Venous (portal, splenic vein) or arterial (1-2% risk).
- Surgical: Bleeding, infection, damage to surrounding structures.
- Other: Reactive thrombocytosis (usually transient).
8.5 Emerging/Experimental Therapies
Under investigation: [1]
- Efgartigimod: Neonatal Fc receptor (FcRn) antagonist - reduces pathogenic IgG (Phase 3 trials ongoing).
- BTK inhibitors: Targeting B cell signalling.
- Complement inhibitors: Sutimlimab.
8.6 Treatment Algorithm Summary
Newly Diagnosed ITP
↓
Count > 30, no bleeding → OBSERVE
↓
Count less than 30 or bleeding → FIRST-LINE
↓
Prednisolone 1mg/kg or Dexamethasone 40mg × 4 days
± IVIg if severe/urgent
↓
Response → Taper steroids → MONITOR
↓
No response / Relapse / Steroid-dependent → SECOND-LINE
↓
TPO-RA (Eltrombopag/Romiplostim) OR Rituximab
↓
Partial response / Intolerance → Alternative TPO-RA / Fostamatinib / Immunosuppressants
↓
Refractory → THIRD-LINE
↓
SPLENECTOMY or Clinical trial
9. Special Situations
9.1 ITP in Pregnancy
Maternal Considerations: [23]
- Platelet count often falls in third trimester (gestational effect).
- Treatment threshold: Treat if count less than 30 × 10⁹/L or less than 50 × 10⁹/L near delivery.
- Safe treatments:
- Corticosteroids: Prednisolone (minimal placental transfer).
- IVIg: Safe and effective.
- Avoid: TPO-RAs (inadequate safety data), rituximab, immunosuppressants.
- Delivery:
- Vaginal delivery safe if maternal count > 50 × 10⁹/L.
- Caesarean section if obstetric indication; platelet count > 50 × 10⁹/L (ideally > 80 × 10⁹/L for neuraxial anaesthesia).
- Platelet transfusion: If required for delivery/haemorrhage.
Neonatal Considerations: [23]
- Neonatal thrombocytopenia: 10-15% of infants (maternal IgG crosses placenta).
- Timing: Nadir platelet count day 2-5 of life.
- Severity: Usually mild; ICH risk less than 1%.
- Management: Check cord blood platelets; monitor for first week; treat neonate if count less than 30 × 10⁹/L (IVIg or transfusion).
9.2 ITP in the Elderly
Specific Challenges: [5]
- Higher bleeding risk: Age-related vascular fragility, comorbidities (hypertension), anticoagulation.
- ICH risk higher: Age > 60 is independent risk factor.
- Treatment tolerance: Greater susceptibility to steroid side effects (diabetes, hypertension, osteoporosis, infection).
- Differential diagnosis: Must exclude MDS (bone marrow examination often indicated).
- Management:
- Lower treatment threshold (treat at higher platelet counts).
- Minimise steroid duration.
- Early use of TPO-RAs.
- Carefully manage comorbidities and concomitant medications.
9.3 Surgery in ITP Patients
Perioperative Management: [1]
- Target platelet count:
- Minor procedures: > 50 × 10⁹/L
- Major surgery / Neuraxial anaesthesia: > 80 × 10⁹/L
- Pre-operative optimisation:
- Corticosteroids (start 1-2 weeks before).
- IVIg (24-48 hours before if urgent rise needed).
- TPO-RAs (weeks in advance; slower onset).
- Intraoperative:
- Meticulous haemostasis.
- Platelet transfusion available if required (with IVIg cover).
- Post-operative: Continue treatment; monitor count daily initially.
9.4 Emergency Bleeding
Life-Threatening Haemorrhage (ICH, major GI bleed): [1,16]
Immediate Management:
- Resuscitation: ABCDE approach, secure airway if reduced GCS.
- Haematology consult: Urgent senior review.
- Platelet transfusion: Despite poor efficacy, transfuse to attempt count > 50 × 10⁹/L.
- IVIg: 1 g/kg IV stat (do not delay).
- High-dose corticosteroids: Methylprednisolone 1g IV or dexamethasone 40mg IV.
- Tranexamic acid: 1g IV (if not contraindicated, e.g., not DIC).
- Imaging: Urgent CT head if ICH suspected.
- Neurosurgical consult: If ICH confirmed.
- Consider: Recombinant Factor VIIa (rFVIIa) in extremis (unlicensed indication; case reports suggest benefit).
Target: Platelet count > 50 × 10⁹/L for haemostasis.
10. Prognosis and Outcomes
10.1 Natural History
Paediatric ITP: [6]
- 80-85% achieve spontaneous remission within 6-12 months.
- Chronic ITP (less than 20%) - persists > 12 months.
- Excellent long-term prognosis.
Adult ITP: [4,5]
- Spontaneous remission: Only 10-20%.
- Chronic course: 60-70% develop chronic ITP (> 12 months).
- Fluctuating counts: Many experience spontaneous fluctuations.
10.2 Treatment Response
First-Line (Corticosteroids): [16]
- Initial response: 70-80%
- Sustained response off treatment: 20-30%
- Relapse common after steroid taper.
Second-Line (TPO-RAs): [18]
- Response rate: 70-80%
- Sustained response on treatment: Majority maintain adequate counts while on therapy.
- Response off treatment: ~30% maintain response after cessation (attempt taper after 3-6 months).
Splenectomy: [22]
- Complete/partial response: 70-80%
- Long-term sustained response (5-10 years): 60-70%
- Relapse: 20-30%
10.3 Mortality
- Overall mortality: Low in most ITP patients (similar to general population).
- Bleeding-related mortality: less than 1-2% overall. [10]
- ICH mortality: 0.5-1% of adult ITP cases; accounts for majority of ITP-related deaths.
- Risk factors for mortality: [10]
- Age > 60 years
- Comorbidities (cardiovascular disease, malignancy)
- Severe refractory ITP (count persistently less than 10 × 10⁹/L)
10.4 Quality of Life
- Chronic ITP significantly impacts quality of life: fatigue, anxiety about bleeding, treatment burden. [24]
- Psychological impact: Fear of bleeding, restrictions on activities (contact sports, travel).
- Treatment side effects: Corticosteroid toxicity major contributor to reduced QoL.
- TPO-RAs: Demonstrated improvements in QoL scores compared to placebo and watch-and-wait. [24]
10.5 Prognosis Scoring
No widely validated prognostic score, but poor prognostic factors include: [5]
- Age > 60 years
- Severe thrombocytopenia at diagnosis (count less than 10 × 10⁹/L)
- Failure to respond to first-line therapy
- Requirement for multiple treatment lines
11. Patient and Layperson Explanation
What is ITP?
Immune Thrombocytopenia (ITP) is a condition where your immune system - which normally protects you from infections - gets confused and attacks your own platelets.
- Platelets are tiny blood cells that help your blood to clot and stop bleeding when you get a cut or injury.
- When you have ITP, your platelet count is low, which means you might bruise easily or bleed more than normal.
Why did I get it?
In most adults with ITP, we don't know exactly why it happens - it is not caused by anything you did or didn't do. Your immune system has mistakenly identified your platelets as "foreign" and is destroying them.
- It is not cancer and it is not contagious.
- In children, ITP often happens after a viral infection and usually goes away on its own.
- In adults, ITP tends to be long-lasting (chronic) and requires monitoring or treatment.
Is it dangerous?
For most people with ITP, the condition is manageable:
- If your platelet count is above 30, you are usually safe to go about normal daily activities.
- If your platelet count is very low (below 10), there is a higher risk of serious bleeding, including bleeding in the brain (intracranial haemorrhage), although this is rare (less than 1%).
Warning signs to seek urgent medical help:
- Blood blisters in your mouth
- Severe headache or confusion
- Vomiting blood or blood in stools
- Severe nosebleeds that won't stop
- Heavy vaginal bleeding
How do we treat it?
The goal of treatment is not to make your platelet count completely normal, but to keep it safe and prevent serious bleeding.
If your count is safe (above 30) and you aren't bleeding:
- We may just watch and wait - regular blood tests to monitor your count.
If treatment is needed:
-
Steroids (tablets like prednisolone):
- Calm down your immune system to stop it attacking your platelets.
- Usually used for a few weeks, then reduced gradually.
-
IVIg (intravenous immunoglobulin):
- Given through a drip in hospital.
- Works quickly to boost your platelet count (useful if you need surgery or are bleeding).
- Effect is temporary (lasts a few weeks).
-
Platelet-boosting medicines (TPO receptor agonists) - e.g., eltrombopag or romiplostim:
- Stimulate your bone marrow to make more platelets.
- Taken long-term as tablets or weekly injections.
- Very effective for most people with chronic ITP.
-
Rituximab:
- An infusion that targets the immune cells making the antibodies against your platelets.
- Can give long-lasting improvement in some people.
-
Surgery (splenectomy) - removing your spleen:
- Your spleen is the main place where platelets are destroyed in ITP.
- Removing it can cure ITP in about 60-70% of people.
- Now less commonly done because medications are so effective.
- Increases your risk of certain infections, so requires vaccinations and lifelong antibiotics.
Can I live a normal life?
Yes, most people with ITP live full, active lives.
- Avoid high-impact contact sports if your count is low (e.g., rugby, boxing).
- Take care to avoid injuries (use electric razors, soft toothbrush).
- Avoid medications that affect platelets (aspirin, ibuprofen) unless specifically advised by your doctor.
- Inform all healthcare providers (dentist, surgeons) about your ITP before any procedures.
- Wear a medical alert bracelet or carry a card with your diagnosis.
Will it go away?
- In children: Yes, usually within 6-12 months.
- In adults: Only about 10-20% have complete remission. Most people will need long-term monitoring, and some will need ongoing treatment.
Questions to ask your doctor
- What is my current platelet count, and what is my target?
- How often do I need blood tests?
- What activities should I avoid?
- What are the side effects of my treatment?
- When should I seek urgent help?
12. References
-
Provan D, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780-3817. doi:10.1182/bloodadvances.2019000812
-
Neunert C, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3(23):3829-3866. doi:10.1182/bloodadvances.2019000966
-
Rodeghiero F, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386-2393. doi:10.1182/blood-2008-07-162503
-
Moulis G, et al. Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood. 2014;124(22):3308-3315. doi:10.1182/blood-2014-05-578336
-
Neunert CE. Management of newly diagnosed immune thrombocytopenia: can we change outcomes? Blood Adv. 2017;1(24):2295-2301. doi:10.1182/bloodadvances.2017009860
-
Neunert C, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117(16):4190-4207. doi:10.1182/blood-2010-08-302984
-
Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002;346(13):995-1008. doi:10.1056/NEJMra010501
-
McMillan R, et al. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood. 2004;103(4):1364-1369. doi:10.1182/blood-2003-08-2672
-
Zufferey A, et al. A paradigm shift in ITP: From autoreactive T cell development to immune dysregulation. Autoimmun Rev. 2017;16(5):511-519. doi:10.1016/j.autrev.2017.03.012
-
Cooper N, Ghanima W. Immune Thrombocytopenia. N Engl J Med. 2019;381(10):945-955. doi:10.1056/NEJMcp1810479
-
Provan D, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115(2):168-186. doi:10.1182/blood-2009-06-225565
-
Stasi R, et al. Effect of Helicobacter pylori eradication on platelet recovery in patients with immune thrombocytopenic purpura: a systematic review. Haematologica. 2009;94(6):850-856. doi:10.3324/haematol.2008.002683
-
Scully M, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312-322. doi:10.1111/jth.13571
-
Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. J Thromb Haemost. 2009;7(6):911-918. doi:10.1111/j.1538-7836.2009.03360.x
-
Greinacher A, et al. Heparin-induced thrombocytopenia. J Thromb Haemost. 2019;17(8):1270-1285. doi:10.1111/jth.14479
-
Mithoowani S, et al. Management of major bleeds in patients with immune thrombocytopenia. J Thromb Haemost. 2020;18(7):1783-1790. doi:10.1111/jth.14863
-
Wei Y, et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood. 2016;127(3):296-302. doi:10.1182/blood-2015-07-659656
-
Kuter DJ, et al. Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: safety and efficacy. Br J Haematol. 2013;161(3):411-423. doi:10.1111/bjh.12260
-
Al-Samkari H, et al. Avatrombopag for the treatment of immune thrombocytopenia. Expert Rev Hematol. 2019;12(10):845-854. doi:10.1080/17474086.2019.1649131
-
Arnold DM, et al. A pilot randomized trial of adjuvant rituximab or placebo for nonsplenectomized patients with immune thrombocytopenia. Blood. 2012;119(6):1356-1362. doi:10.1182/blood-2011-08-374777
-
Bussel J, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am J Hematol. 2018;93(7):921-930. doi:10.1002/ajh.25125
-
Vianelli N, et al. Splenectomy as a curative treatment for immune thrombocytopenia: a retrospective analysis of 233 patients with a minimum follow up of 10 years. Haematologica. 2013;98(6):875-880. doi:10.3324/haematol.2012.075648
-
Gernsheimer T, et al. How I treat thrombocytopenia in pregnancy. Blood. 2013;121(1):38-47. doi:10.1182/blood-2012-08-448944
-
McMillan R, Bussel JB, George JN, Lalla D, Nichol JL. Self-reported health-related quality of life in adults with chronic immune thrombocytopenic purpura. Am J Hematol. 2008;83(2):150-154. doi:10.1002/ajh.21123
Document Information
- Last Updated: 2026-01-05
- Version: 2.0 (Enhanced)
- Word Count: ~7,800 words
- Target Audience: Medical students, trainee haematologists, MRCP/FRACP candidates
- Citation Count: 24 PubMed-indexed references
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Haematopoiesis and Platelet Production
- Coagulation Cascade
Differentials
Competing diagnoses and look-alikes to compare.
- Thrombotic Thrombocytopenic Purpura (TTP)
- Disseminated Intravascular Coagulation (DIC)
- Heparin-Induced Thrombocytopenia (HIT)
- Aplastic Anaemia
Consequences
Complications and downstream problems to keep in mind.
- Intracranial Haemorrhage
- Post-Splenectomy Sepsis