Myelodysplastic Syndromes
The diagnosis of MDS requires bone marrow examination demonstrating dysplastic changes in ≥10% of cells in one or more myeloid lineages, with exclusion of other causes of dysplasia including nutritional deficiencies...
What matters first
The diagnosis of MDS requires bone marrow examination demonstrating dysplastic changes in ≥10% of cells in one or more myeloid lineages, with exclusion of other causes of dysplasia including nutritional deficiencies...
Severe symptomatic anaemia (Hb less than 70 g/L)
9 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Severe symptomatic anaemia (Hb less than 70 g/L)
- Neutropenic sepsis (ANC less than 0.5 with fever)
- Severe thrombocytopenia with active bleeding (platelets less than 10)
- Transformation to AML (blasts >=20%)
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Myelodysplastic Syndromes
1. Clinical Overview
Summary
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal haematopoietic stem cell disorders characterised by ineffective haematopoiesis leading to peripheral blood cytopenias despite a normocellular or hypercellular bone marrow, morphological dysplasia in one or more myeloid cell lineages, and an inherent risk of progression to acute myeloid leukaemia (AML). MDS predominantly affects older adults with a median age at diagnosis of 70-75 years and represents the most common haematological malignancy in this age group. [1] The hallmark of MDS is the paradox of bone marrow hypercellularity with peripheral cytopenias, resulting from increased intramedullary apoptosis of haematopoietic precursors. Macrocytic anaemia is the most common presenting feature, occurring in approximately 80-85% of patients at diagnosis. [2]
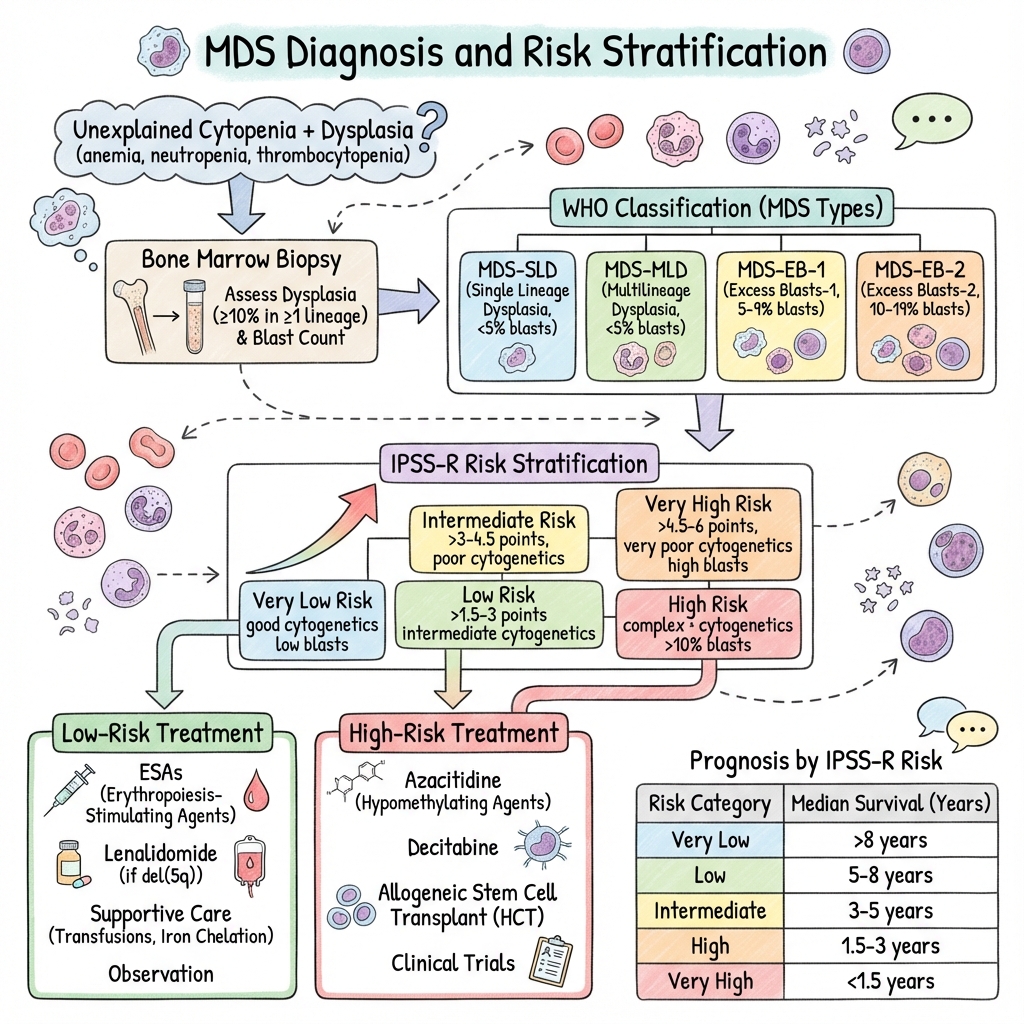
The diagnosis of MDS requires bone marrow examination demonstrating dysplastic changes in ≥10% of cells in one or more myeloid lineages, with exclusion of other causes of dysplasia including nutritional deficiencies (vitamin B12, folate, copper), recent cytotoxic therapy, and congenital disorders. [3] Risk stratification using the Revised International Prognostic Scoring System (IPSS-R) is essential for therapeutic decision-making, as it incorporates cytogenetic abnormalities, bone marrow blast percentage, and depth of cytopenias to categorize patients into five risk categories with markedly different survival and AML transformation rates. [4]
Treatment strategy is fundamentally determined by IPSS-R risk category: lower-risk MDS (Very Low, Low, and some Intermediate risk) is managed primarily with supportive care including red cell transfusions, erythropoiesis-stimulating agents (ESAs), and targeted therapies such as lenalidomide for del(5q) MDS or luspatercept for ring sideroblast MDS. [5,6] Higher-risk MDS (Intermediate, High, and Very High risk) requires disease-modifying therapy with hypomethylating agents (azacitidine or decitabine), with allogeneic haematopoietic stem cell transplantation (allo-HSCT) representing the only potentially curative therapeutic option for eligible patients. [7,8]
The natural history of MDS is characterized by progressive bone marrow failure with worsening cytopenias, increasing transfusion dependence, and evolution to AML in approximately 30% of cases overall, though this risk varies dramatically by subtype from less than 5% in isolated del(5q) MDS to > 80% in MDS with excess blasts-2. [9] Molecular characterization has revealed recurrent mutations in genes regulating RNA splicing (SF3B1, SRSF2, U2AF1), DNA methylation (TET2, DNMT3A, IDH1/2), chromatin modification (ASXL1, EZH2), and tumour suppression (TP53), which have important diagnostic, prognostic, and increasingly therapeutic implications. [10]
Key Facts
- Definition: Clonal haematopoietic stem cell disorders with ineffective haematopoiesis, dysplasia (≥10% in ≥1 lineage), cytopenias, and AML transformation risk
- Incidence: 4-5 per 100,000 per year overall; increases to > 30 per 100,000 per year in adults aged > 70 years [1]
- Peak Demographics: Median age 70-75 years; male predominance (M:F ratio 1.5-2:1) [2]
- AML Transformation: Cumulative incidence 30% overall; varies by subtype (5% in del(5q) to 80% in MDS-EB-2) [9]
- Gold Standard Investigation: Bone marrow aspirate and trephine biopsy with morphological assessment, cytogenetics, FISH, and molecular profiling
- Risk Stratification: IPSS-R incorporating cytogenetics (5 categories), BM blasts, Hb, platelets, ANC (5 risk groups: Very Low to Very High) [4]
- First-line Treatment Lower-Risk: Supportive care, ESAs (if EPO less than 500 mU/mL), lenalidomide (del(5q)), luspatercept (ring sideroblasts) [5,6]
- First-line Treatment Higher-Risk: Azacitidine 75 mg/m² SC days 1-7 q28d, or allo-HSCT in eligible patients [7,8]
- Prognosis: Median survival ranges from 8.8 years (Very Low risk) to 0.8 years (Very High risk) by IPSS-R [4]
- Molecular Landscape: Mutations in > 40 genes; most common: SF3B1 (20-25%), TET2 (20-25%), ASXL1 (15-20%), SRSF2 (10-15%), DNMT3A (10-15%), RUNX1 (10%), TP53 (5-10%) [10]
Clinical Pearls
Diagnostic Pearl: MDS cannot be diagnosed from peripheral blood alone—bone marrow examination is mandatory. Dysplasia must involve ≥10% of cells in at least one myeloid lineage. Single-lineage cytopenias with less than 10% dysplasia may represent clonal cytopenias of indeterminate potential (CCUS) or idiopathic cytopenias of undetermined significance (ICUS). [3]
Treatment Pearl: Erythropoietin level is the strongest predictor of response to ESAs in lower-risk MDS. Patients with baseline EPO less than 500 mU/mL have 60-70% response rates, while those with EPO > 500 mU/mL have less than 10% response rates. Combining EPO with G-CSF may improve response in selected patients. [11]
Cytogenetic Pearl: The presence of isolated del(5q) defines a distinct MDS subtype with favourable prognosis (median survival > 5 years) and high responsiveness to lenalidomide (67% transfusion independence rate, 45% complete cytogenetic response). [5]
Molecular Pearl: SF3B1 mutations are highly associated with ring sideroblasts (> 90% of cases) and predict favourable prognosis and excellent response to luspatercept. Conversely, TP53 mutations (especially multi-hit) confer very poor prognosis with median survival less than 12 months and poor transplant outcomes. [10,12]
Transfusion Pearl: Each unit of red blood cells contains 200-250 mg of iron. Patients receiving > 20-25 units develop clinically significant iron overload with cardiac and hepatic complications. Initiate iron chelation therapy when ferritin exceeds 1000 μg/L in transfusion-dependent patients with life expectancy > 1 year. [13]
Pitfall Warning: Always exclude nutritional causes before diagnosing MDS. Vitamin B12 deficiency, folate deficiency, and copper deficiency can all produce macrocytic anaemia with dysplastic morphology. Heavy alcohol use, HIV infection, and recent chemotherapy can also cause reactive dysplasia. [3]
Transplant Pearl: The timing of allo-HSCT in MDS is controversial. Some advocate early transplant in higher-risk disease, while others recommend delaying until disease progression to maximize quality-adjusted life years. IPSS-R Very High and High risk patients generally benefit from early transplant, while Intermediate risk is more nuanced. [14]
Mnemonic - MDS Features: D-R-I-C-H - Dysplasia (morphological), Risk stratification (IPSS-R), Ineffective haematopoiesis, Cytopenias (peripheral), Hypercellular marrow
Mnemonic - IPSS-R Components: C-B-H-P-N - Cytogenetics, Blasts, Haemoglobin, Platelets, Neutrophils (ANC)
Why This Matters Clinically
MDS represents the most common haematological malignancy in adults over 70 years, with incidence rising dramatically in ageing populations. Accurate risk stratification using IPSS-R fundamentally determines treatment intensity and dramatically impacts outcomes—treating lower-risk patients with intensive therapy causes toxicity without benefit, while undertreating higher-risk patients accelerates progression to AML. The emergence of targeted therapies (lenalidomide for del(5q), luspatercept for ring sideroblasts) and improved transplant outcomes has transformed management over the past decade. Early recognition and referral to haematology is critical, as delayed diagnosis leads to transfusion dependence and iron overload complications. Molecular profiling increasingly guides prognosis and therapy selection, with implications for clinical trial eligibility and emerging targeted agents.
2. Epidemiology
Incidence and Prevalence
Age-Adjusted Incidence: [1,2]
- Overall population: 4-5 cases per 100,000 person-years
- Age 60-69 years: 15-20 per 100,000 per year
- Age 70-79 years: 30-40 per 100,000 per year
- Age ≥80 years: 50-60 per 100,000 per year
- Prevalence estimated at 60,000-170,000 in the United States
Incidence Trends:
The reported incidence of MDS has increased 2-3 fold over the past 30 years, attributed to:
- Ageing population demographics
- Improved diagnostic recognition and classification
- Increased bone marrow examination rates in elderly patients with unexplained cytopenias
- Survival from prior malignancies treated with chemotherapy/radiotherapy
- Heightened clinical awareness
Demographics
| Factor | Distribution | Details |
|---|---|---|
| Age | Median 70-75 years | Rare before age 50 (less than 10% of cases); incidence rises exponentially after age 60 |
| Sex | M:F ratio 1.5-2:1 | Male predominance across all age groups and subtypes |
| Ethnicity | Slight variations | Highest rates in white populations; lower in Asian populations; similar rates in Hispanic and African populations |
| Geography | Western > Eastern | Higher reported incidence in Europe and North America; may reflect diagnostic access |
Risk Factors and Causation
Established Risk Factors: [15]
| Risk Factor | Relative Risk | Mechanism | Details |
|---|---|---|---|
| Advanced age | Exponential increase | Accumulation of somatic mutations, clonal haematopoiesis | Risk increases 10-fold from age 60 to 80 |
| Prior chemotherapy | 5-20x | DNA damage, chromosomal instability | Alkylating agents (cyclophosphamide, melphalan): 5-7 year latency, often del(5q)/del(7q); Topoisomerase II inhibitors (etoposide, anthracyclines): 1-3 year latency, often 11q23/MLL rearrangements |
| Prior radiotherapy | 3-10x | Ionizing radiation-induced mutations | Dose-dependent; field-dependent (pelvic > other sites) |
| Occupational benzene | 2-5x | Haematopoietic stem cell damage | Dose-dependent; historical exposure in chemical, petroleum, rubber industries |
| Cigarette smoking | 1.5-2x | Benzene and other carcinogens | Active smoking; dose-dependent relationship |
| Autoimmune disease | 1.5-3x | Chronic immune stimulation | Especially rheumatoid arthritis, systemic lupus |
Therapy-Related MDS (t-MDS): [15]
- Accounts for 10-15% of all MDS cases
- Median latency: 5-7 years after alkylating agents; 1-3 years after topoisomerase II inhibitors
- Typically presents as higher-risk disease with complex karyotype
- Poor prognosis: median survival 8-10 months; high early AML transformation
- Most common after treatment for breast cancer, lymphoma, ovarian cancer, testicular cancer
Genetic Predisposition Syndromes:
- Fanconi anaemia (DNA repair defect)
- Dyskeratosis congenita (telomere maintenance)
- Shwachman-Diamond syndrome (ribosomal dysfunction)
- Diamond-Blackfan anaemia
- Familial platelet disorder with propensity to AML (RUNX1 germline)
- GATA2 deficiency
- DDX41 germline mutations (later-onset familial MDS)
Clonal Haematopoiesis of Indeterminate Potential (CHIP): [16]
- Defined as presence of somatic mutations (VAF ≥2%) in haematopoietic stem cells without cytopenias or dysplasia
- Present in 10-15% of individuals > 70 years
- Confers 0.5-1% annual risk of progression to MDS/AML
- Most common mutations: DNMT3A, TET2, ASXL1 ("DTA" mutations)
- Risk factors for progression: VAF > 10%, multiple mutations, TP53/spliceosome mutations
3. Pathophysiology
Molecular Pathogenesis: The Multi-Hit Model
Stage 1: Initiating Mutations and Clonal Expansion [10,16]
MDS arises from the stepwise acquisition of somatic mutations in haematopoietic stem cells, leading to clonal dominance:
-
Early Events (Age-related/CHIP):
- DNMT3A, TET2, ASXL1 mutations confer proliferative advantage
- Mutated clones expand gradually over years/decades
- Present in 10-15% of healthy elderly individuals
- Pre-malignant state with normal blood counts
-
Disease-Defining Mutations:
- Additional mutations in splicing factors (SF3B1, SRSF2, U2AF1), signalling (NRAS, KRAS, JAK2), or transcription factors (RUNX1, ETV6)
- "Threshold effect: typically 2-3+ mutations required for MDS phenotype"
- Clonal fraction expands to > 10% of haematopoiesis
Stage 2: Ineffective Haematopoiesis and Dysplasia [2,10]
The characteristic features of MDS result from multiple converging mechanisms:
Mechanisms of Cytopenias:
-
Increased Intramedullary Apoptosis (dominant in lower-risk MDS):
- Activated death receptor pathways (Fas/FasL)
- Mitochondrial dysfunction
- Pro-apoptotic cytokine milieu (TNF-α, IFN-γ)
- Result: Hypercellular marrow with peripheral cytopenias ("ineffective haematopoiesis")
-
Aberrant Differentiation:
- Dysplastic morphology: nuclear-cytoplasmic asynchrony, abnormal granulation, multinucleation
- Functional defects: defective erythrocyte membrane, hypogranular neutrophils, dysfunctional platelets
-
Immune Dysregulation:
- T-cell mediated suppression of normal haematopoiesis
- Inflammatory microenvironment
- May explain response to immunosuppression in select cases
Stage 3: Clonal Evolution and Disease Progression [9,17]
Higher-risk MDS and transformation to AML involves:
-
Acquisition of Additional Mutations:
- Second hits in signalling pathways (FLT3, NRAS, KIT)
- TP53 mutations (associated with complex karyotype, poor prognosis)
- Loss of tumour suppressors
- Expansion of subclones with competitive advantage
-
Shift from Apoptosis to Proliferation:
- Decreased apoptosis in blast population
- Increased self-renewal capacity
- Resistance to differentiation signals
-
Blast Accumulation:
- 5-9% blasts: MDS-EB-1 (Excess Blasts-1)
- 10-19% blasts: MDS-EB-2 (Excess Blasts-2)
- ≥20% blasts: AML by definition
Recurrent Cytogenetic Abnormalities
Prognostic Classification (IPSS-R Cytogenetic Risk Groups): [4]
| Risk Category | Chromosomal Abnormalities | Frequency | Prognosis |
|---|---|---|---|
| Very Good | -Y, del(11q) | 3-5% | Favourable |
| Good | Normal karyotype, del(5q) alone, del(12p), del(20q), double including del(5q) | 65-70% | Intermediate-favourable |
| Intermediate | del(7q), +8, +19, i(17q), other single/double | 10-15% | Intermediate |
| Poor | -7, inv(3)/t(3q)/del(3q), double including -7/del(7q), complex (3 abnormalities) | 10-12% | Poor |
| Very Poor | Complex (> 3 abnormalities) | 8-10% | Very poor |
Key Cytogenetic Entities:
- Isolated del(5q): Deletion of 5q31-33; associated with lenalidomide responsiveness; median survival > 5 years [5]
- Monosomy 7/del(7q): Poor prognosis; common in therapy-related MDS
- Complex karyotype (≥3 abnormalities): Very poor prognosis; often associated with TP53 mutation; median survival 9-12 months
- Normal karyotype: Most common (50-60%); intermediate prognosis; molecular profiling critical for risk stratification
Recurrent Molecular Mutations and Functional Categories
1. RNA Splicing Machinery (25-30% of MDS): [10]
- SF3B1 (20-25%): Strongly associated with ring sideroblasts (> 90%); favourable prognosis; luspatercept-responsive
- SRSF2 (10-15%): Associated with multilineage dysplasia, thrombocytosis; poor prognosis
- U2AF1 (5-10%): Associated with ring sideroblasts, thrombocytopenia; intermediate prognosis
- ZRSR2 (5%): X-linked; mild phenotype
2. DNA Methylation (40-50% of MDS):
- TET2 (20-25%): Loss of function; associated with CHIP; may predict hypomethylating agent response
- DNMT3A (10-15%): Common in CHIP; intermediate prognosis
- IDH1/IDH2 (5-10%): Produce oncometabolite 2-HG; targetable with IDH inhibitors
3. Chromatin Modification (20-30% of MDS):
- ASXL1 (15-20%): Loss of function; poor prognosis; associated with higher-risk disease
- EZH2 (5%): Loss of function; poor prognosis
4. Transcription Factors (10-15% of MDS):
- RUNX1 (10%): Poor prognosis; higher risk of AML transformation
- ETV6 (3-5%): Associated with thrombocytopenia
5. Tumour Suppressors:
- TP53 (5-10%): Very poor prognosis; multi-hit TP53 (biallelic) worse than mono-allelic; median survival 9-12 months; poor transplant outcomes; associated with complex karyotype and therapy-related MDS [12]
6. Signalling Pathways (5-10%):
- NRAS/KRAS (5-10%): Late events; associated with progression
- JAK2 (3-5%): V617F mutation; overlap with MPN
- FLT3 (less than 5%): More common in transformation to AML
7. Cohesin Complex (10-15%):
- STAG2, RAD21, SMC1A: Associated with higher-risk disease
Classification Systems
WHO Classification (5th Edition, 2022): [3]
| Subtype | Abbreviation | Dysplasia | Blasts (BM) | Ring Sideroblasts | Key Features |
|---|---|---|---|---|---|
| MDS with low blasts and isolated del(5q) | MDS-del(5q) | Variable | less than 5% | Any | Isolated del(5q) or with 1 additional abnormality except -7/del(7q); lenalidomide-responsive |
| MDS with low blasts | MDS-LB | ≥10% in 1+ lineage | less than 5% | less than 15% or 5-14% without SF3B1 | Formerly MDS-SLD/MDS-MLD |
| MDS with ring sideroblasts | MDS-RS | ≥10% in 1+ lineage | less than 5% | ≥15% or ≥5% with SF3B1 mutation | Luspatercept-responsive; good prognosis |
| MDS with increased blasts | MDS-IB | Any | 5-19% | Any | Subdivided into MDS-IB1 (5-9%) and MDS-IB2 (10-19%); higher risk |
| MDS, hypoplastic | MDS-h | ≥10% in 1+ lineage | less than 5-19% | Any | Hypocellular BM (less than 25% cellularity); DDx with aplastic anaemia |
| MDS with fibrosis | MDS-f | ≥10% in 1+ lineage | less than 5-19% | Any | MF-2 or MF-3 fibrosis; poor prognosis |
| MDS, unclassifiable | MDS-U | Variants | less than 5% | Any | Does not fit other categories |
International Consensus Classification (ICC, 2022):
Similar to WHO 5th edition with minor terminology differences; emphasizes molecular genetics.
4. Clinical Presentation
Symptom Profile
At Diagnosis: [2]
- Asymptomatic (20-30%): Incidental finding on routine blood tests
- Symptomatic anaemia (60-70%): Most common presenting feature
- Infection-related (10-15%): Due to neutropenia
- Bleeding-related (5-10%): Due to thrombocytopenia
- Constitutional symptoms (10-20%): Fatigue, weight loss (more common in higher-risk disease)
Anaemia-Related Symptoms (most common):
- Fatigue and reduced exercise tolerance (85-90%)
- Exertional dyspnoea (60-70%)
- Dizziness, lightheadedness (40-50%)
- Pallor (visible when Hb less than 100 g/L)
- Palpitations, chest discomfort (30-40%, especially in elderly with cardiovascular comorbidity)
- Cognitive impairment in elderly (may be subtle)
Neutropenia-Related Symptoms (10-20%):
- Recurrent bacterial infections (skin, respiratory, urinary)
- Oral ulceration (aphthous-like ulcers)
- Perianal infections
- Severe infections when ANC less than 0.5 × 10⁹/L
Thrombocytopenia-Related Symptoms (10-15%):
- Easy bruising, ecchymoses
- Petechiae (typically platelets less than 50 × 10⁹/L)
- Mucosal bleeding (gingival, epistaxis)
- Menorrhagia (in premenopausal women, rare in MDS demographics)
- Severe bleeding when platelets less than 10 × 10⁹/L
Constitutional Symptoms (more common in higher-risk MDS):
- Unintentional weight loss
- Low-grade fever (exclude infection)
- Night sweats
- Bone pain (uncommon, consider transformation if present)
Clinical Examination Findings
General Appearance:
- Pallor (conjunctival, palmar)
- Cachexia or frailty (higher-risk disease)
- Petechial rash (trunk, lower extremities)
- Ecchymoses at venepuncture sites or from minor trauma
Abdominal Examination:
- Splenomegaly: Present in less than 10% of MDS cases
- "Mild splenomegaly (1-2 cm below costal margin): May occur in MDS with ring sideroblasts or associated with JAK2 mutation (MDS/MPN overlap)"
- "Moderate-to-marked splenomegaly: Should prompt consideration of MDS/MPN overlap syndromes (CMML, MDS/MPN-U) or transformation to AML"
- Hepatomegaly: Rare in uncomplicated MDS; consider iron overload if long-term transfusion-dependent
Lymphatic Examination:
- Lymphadenopathy: Typically absent in MDS
- "If present, consider: (1) Concurrent infection, (2) Transformation to AML, (3) Alternative diagnosis (lymphoma, CMML)"
Oropharyngeal Examination:
- Oral candidiasis (neutropenia)
- Aphthous-like ulcers (neutropenia)
- Gingival bleeding (thrombocytopenia)
Skin Examination:
- Petechiae (non-blanching, 1-2 mm)
- Purpura (3-10 mm)
- Ecchymoses (> 1 cm)
- Skin infections (cellulitis, abscesses if neutropenic)
Cardiovascular Examination:
- Tachycardia (compensation for anaemia)
- Flow murmur (anaemia)
- Signs of high-output cardiac failure in severe chronic anaemia
Red Flag Examination Findings:
[!CAUTION]
- Moderate-to-marked splenomegaly: Suggests MDS/MPN overlap or transformation
- Lymphadenopathy: Atypical for MDS; investigate alternative causes
- Skin infiltration (leukaemia cutis): Suggests transformation to AML
- Bone tenderness: Consider transformation or alternative diagnosis
- Neurological signs: Rare; consider paraneoplastic phenomena or coincidental pathology
5. Investigations
Diagnostic Pathway
Step 1: First-Line Blood Tests (for unexplained cytopenia)
| Test | Expected Findings in MDS | Purpose |
|---|---|---|
| Full blood count | Anaemia (80-85%), neutropenia (40-50%), thrombocytopenia (40-50%); isolated or combined | Identify cytopenias |
| Blood film | Macrocytosis (MCV 100-115 fL), anisopoikilocytosis, oval macrocytes, hypogranular neutrophils, pseudo-Pelger-Huët cells, hypolobulated megakaryocytes | Morphological dysplasia |
| Reticulocyte count | Inappropriately low or normal for degree of anaemia (typically less than 100 × 10⁹/L) | Confirms ineffective erythropoiesis |
| Vitamin B12 | Normal (rule out deficiency) | Exclude B12 deficiency (causes macrocytosis + dysplasia) |
| Folate (red cell) | Normal (rule out deficiency) | Exclude folate deficiency |
| Copper/caeruloplasmin | Normal (if history of malabsorption or zinc excess) | Exclude copper deficiency (rare mimic) |
| TSH | Normal (rule out hypothyroidism) | Exclude thyroid cause of macrocytosis |
| LDH | Often elevated (due to ineffective haematopoiesis and intramedullary cell death) | Non-specific; marker of cell turnover |
| EPO level | Variable; prognostic for ESA response | Predict ESA responsiveness (less than 500 mU/mL favourable) [11] |
| Ferritin | May be elevated (dysplastic erythropoiesis, transfusions) | Assess iron stores and overload risk |
| HIV, Hepatitis B/C | Screen in at-risk individuals | Exclude viral causes of cytopenias |
Peripheral Blood Film Dysplastic Features:
- Erythroid: Macrocytosis, oval macrocytes, anisopoikilocytosis, basophilic stippling, nucleated red cells
- Myeloid: Hypogranular neutrophils, pseudo-Pelger-Huët anomaly (bilobed nuclei), agranular neutrophils
- Megakaryocytic: Circulating micromegakaryocytes, hypolobulated platelets, giant platelets
Step 2: Bone Marrow Examination (MANDATORY for MDS diagnosis) [3]
| Component | Technique | Information Obtained |
|---|---|---|
| Aspirate | Liquid sample for morphology | Blast count (500-cell differential), dysplasia assessment (% dysplastic cells per lineage), iron stores, ring sideroblast count |
| Trephine biopsy | Core tissue sample | Cellularity (hypo/normo/hypercellular), fibrosis (reticulin/collagen grading MF 0-3), architecture, focal lesions |
| Cytogenetics (karyotype) | Metaphase chromosome analysis | Clonal abnormalities (del(5q), -7/del(7q), +8, complex karyotype, etc.); IPSS-R prognostic category [4] |
| FISH | Targeted probe analysis | Detect specific abnormalities (del(5q), del(7q), del(20q), trisomy 8, del(17p)) when metaphases inadequate |
| Flow cytometry | Surface/intracellular markers | Blast immunophenotype, aberrant antigen expression, exclude other diagnoses |
| Molecular testing (NGS) | Next-generation sequencing panel | Mutations in SF3B1, TP53, ASXL1, TET2, SRSF2, RUNX1, DNMT3A, etc.; prognostic and therapeutic implications [10] |
| Iron stain (Perls) | Prussian blue stain on aspirate | Ring sideroblasts (≥5 siderotic granules encircling ≥1/3 of nucleus); diagnose MDS-RS |
Dysplasia Criteria (WHO 2022): [3]
Dysplasia must involve ≥10% of cells in at least one myeloid lineage:
- Erythroid dysplasia: Nuclear budding, internuclear bridging, multinuclearity, karyorrhexis, megaloblastic changes, PAS-positive granules, ring sideroblasts
- Myeloid dysplasia: Hypogranularity, pseudo-Pelger-Huët anomaly, hypersegmentation, abnormal chromatin clumping
- Megakaryocytic dysplasia: Micromegakaryocytes, hypolobulated nuclei (mono/bilobed), multiple separated nuclei
Blast Count:
- Count on 500-cell bone marrow differential
- Blasts include myeloblasts, monoblasts, and promonocytes
- Erythroid precursors are excluded from denominator unless erythroid less than 50%
- less than 5% blasts: Low-risk MDS
- 5-9% blasts: MDS-IB1
- 10-19% blasts: MDS-IB2
- ≥20% blasts: AML (no longer MDS)
Ring Sideroblasts:
- ≥5 siderotic granules encircling ≥1/3 of nuclear circumference
- Seen on Perls' Prussian blue stain
- ≥15% ring sideroblasts (or ≥5% with SF3B1 mutation): Defines MDS-RS subtype
Step 3: Risk Stratification - IPSS-R [4]
The Revised International Prognostic Scoring System (IPSS-R) is the gold standard for MDS risk stratification and incorporates:
IPSS-R Variables and Scoring:
| Variable | Points |
|---|---|
| Cytogenetics | Very good (0), Good (1), Intermediate (2), Poor (3), Very poor (4) |
| BM Blasts % | ≤2% (0), > 2-less than 5% (1), 5-10% (2), > 10% (3) |
| Haemoglobin | ≥10 g/dL (0), 8-less than 10 (1), less than 8 (1.5) |
| Platelets | ≥100 (0), 50-less than 100 (0.5), less than 50 (1) |
| ANC | ≥0.8 (0), less than 0.8 (0.5) |
IPSS-R Risk Categories and Outcomes:
| Risk Category | Score | Median OS (years) | Median Time to 25% AML (years) | % of Patients |
|---|---|---|---|---|
| Very Low | ≤1.5 | 8.8 | Not reached | 20% |
| Low | > 1.5-3 | 5.3 | 10.8 | 35% |
| Intermediate | > 3-4.5 | 3.0 | 3.2 | 20% |
| High | > 4.5-6 | 1.6 | 1.4 | 15% |
| Very High | > 6 | 0.8 | 0.7 | 10% |
Treatment Implications:
- Lower-Risk MDS (Very Low, Low, Intermediate in selected patients): Supportive care, ESAs, lenalidomide (del(5q)), luspatercept (RS)
- Higher-Risk MDS (Intermediate/High/Very High): Hypomethylating agents, allo-HSCT consideration
Step 4: Additional Molecular Risk Stratification
Recent data support integration of molecular mutations into prognostic models: [10,12]
Very High-Risk Molecular Features:
- TP53 multi-hit (biallelic mutation or mutation + del(17p)): Median OS 9-12 months; poor transplant outcomes
- TP53 mono-allelic (single mutation, VAF > 10%): Intermediate-poor prognosis
- ASXL1, RUNX1, EZH2 in combination: Additive poor prognosis
Favourable Molecular Features:
- SF3B1 mutation (especially without high-risk co-mutations): Good prognosis; luspatercept response [6]
- Isolated del(5q): Excellent prognosis; lenalidomide response [5]
Emerging Prognostic Models:
- IPSS-M (Molecular IPSS): Incorporates 31 genes; under validation
- MDS-specific gene panels increasingly standard in clinical practice
6. Differential Diagnosis
Conditions Mimicking MDS:
| Condition | Distinguishing Features |
|---|---|
| Vitamin B12 deficiency | Elevated methylmalonic acid and homocysteine; low B12; macrocytosis may be marked (MCV > 110); hypersegmented neutrophils common; reversible with B12 replacement |
| Folate deficiency | Low red cell folate; macrocytosis; dietary history; alcohol excess; reversible |
| Copper deficiency | History of gastric surgery, zinc excess, malabsorption; low copper/caeruloplasmin; vacuolated myeloid precursors; neuropathy common |
| Aplastic anaemia | Hypocellular marrow (less than 25%); no dysplasia; pancytopenia without dysplastic features; may respond to immunosuppression |
| Paroxysmal nocturnal haemoglobinuria (PNH) | Flow cytometry positive for PNH clone (CD55/CD59 deficiency); haemolysis; may coexist with MDS |
| HIV infection | Positive serology; cytopenias multifactorial; dysplasia may be reactive |
| Alcohol excess | Macrocytosis often marked; thrombocytopenia; vacuolated erythroid precursors; reversible with abstinence |
| Chronic liver disease | Macrocytosis; thrombocytopenia (hypersplenism); dysplastic-appearing megakaryocytes; no clonal abnormality |
| Recent chemotherapy | Temporal relationship; reactive dysplasia; recovery expected within 3-6 months |
| Hypothyroidism | Elevated TSH; macrocytosis; reversible with thyroid replacement |
| Congenital dyserythropoietic anaemia | Onset in childhood/young adulthood; specific morphological features; family history |
| AML with low blast count | Recurrent genetic abnormalities (t(8;21), inv(16), t(15;17)) define AML even if blasts less than 20%; different treatment |
MDS vs. MDS/MPN Overlap Syndromes:
| Feature | MDS | CMML | MDS/MPN-U |
|---|---|---|---|
| Monocytes | less than 1 × 10⁹/L | ≥1 × 10⁹/L (> 10% of WBC) | less than 1 × 10⁹/L |
| Dysplasia | Present | Present | Present |
| Proliferative features | Absent | May have mild splenomegaly, leukocytosis | Present (WBC > 13, splenomegaly) |
| Blasts | less than 20% | less than 20% | less than 20% |
7. Management
Treatment Strategy by Risk Category
Fundamental Principle: Treatment intensity is determined by IPSS-R risk category, not patient age alone. [7,8,18]
Lower-Risk MDS (IPSS-R Very Low / Low / Selected Intermediate)
Goals: Improve cytopenias, reduce transfusion burden, improve quality of life, delay progression
1. Watchful Waiting
- Indication: Asymptomatic patients with mild, stable cytopenias
- Monitoring: FBC every 3-6 months; bone marrow examination if progressive cytopenias or increasing blasts
2. Erythropoiesis-Stimulating Agents (ESAs) [11,18]
- Indication: Symptomatic anaemia (Hb typically less than 100 g/L), IPSS-R Very Low/Low/Int risk, transfusion-independent or low burden (less than 2 units/month)
- Predictors of Response: EPO less than 500 mU/mL (60-70% response), transfusion independence or low burden, lower BM blast %
- Agents:
- "Erythropoietin alfa or beta: 30,000-60,000 units SC weekly (or 150-300 units/kg 3x/week)"
- "Darbepoetin alfa: 150-300 μg SC every 1-2 weeks"
- Response Assessment: After 8-12 weeks; response = Hb increase ≥15 g/L or transfusion independence
- Duration: Continue if responding; discontinue if no response by 12 weeks
- ± G-CSF: May improve erythroid response rates by 20-30% when combined with ESA
3. Lenalidomide (del(5q) MDS) [5,18]
- Indication: MDS with del(5q) with or without additional abnormalities (except -7/del(7q)), transfusion-dependent anaemia
- Dose: 10 mg PO daily days 1-21 of 28-day cycle (or continuous daily)
- Efficacy: 67% achieve transfusion independence; 45% achieve complete cytogenetic response (loss of del(5q) clone); median duration of transfusion independence 2+ years
- Toxicity: Myelosuppression (80% grade 3-4 neutropenia, 60% thrombocytopenia) requiring dose reductions/interruptions; DVT/PE risk (5-10%, consider thromboprophylaxis)
- Monitoring: FBC weekly × 8 weeks, then every 2-4 weeks; dose adjust for cytopenias
- Predictors of Response: Isolated del(5q) better than del(5q) + 1 additional abnormality; transfusion burden less than 4 units/month; TP53 wild-type
4. Luspatercept (MDS-RS) [6,18]
- Indication: MDS with ring sideroblasts (≥15% RS or ≥5% with SF3B1 mutation), IPSS-R Very Low/Low/Intermediate risk, transfusion-dependent anaemia, failed ESA or EPO > 200 mU/mL
- Mechanism: Activin receptor ligand trap; promotes late-stage erythropoiesis
- Dose: 1.0 mg/kg SC every 3 weeks; escalate to 1.33 and 1.75 mg/kg if inadequate response
- Efficacy: 38% achieve transfusion independence ≥8 weeks (vs 13% placebo); 53% achieve ≥50% transfusion reduction
- Predictors of Response: SF3B1 mutation, baseline EPO less than 200 mU/mL, baseline transfusion burden less than 6 units/8 weeks
- Toxicity: Generally well-tolerated; fatigue, bone pain, hypertension (monitor BP)
5. Immunosuppressive Therapy (Selected Cases) [18]
- Indication: Hypocellular MDS (less than 30% BM cellularity), younger age (less than 60 years), HLA-DR15 positivity, trisomy 8, short disease duration, PNH clone
- Agents: Antithymocyte globulin (ATG) + ciclosporin (similar to aplastic anaemia protocols)
- Response: 30-40% haematological response in selected patients
- Consider: When overlap with aplastic anaemia suspected
6. Red Cell Transfusion Support
- Indication: Symptomatic anaemia unresponsive to ESA/lenalidomide/luspatercept
- Target: Hb 80-100 g/L (individualize based on symptoms, comorbidities)
- Strategy: Transfuse to maintain functional capacity; avoid over-transfusion
7. Iron Chelation Therapy [13,18]
- Rationale: Each unit of RBC contains 200-250 mg iron; transfusion-dependent patients accumulate 0.3-0.5 mg/kg/day iron excess
- Indication: Transfusion-dependent (typically > 20-25 units lifetime), ferritin > 1000 μg/L, life expectancy > 1 year, IPSS-R Very Low/Low risk
- Goal: Prevent cardiac and hepatic iron toxicity
- Agents:
- "Deferasirox: 20-30 mg/kg PO daily (first-line); monitor renal function, LFTs"
- "Deferoxamine: 25-50 mg/kg SC infusion 5-7 days/week (second-line; cumbersome administration)"
- "Deferiprone: 75-100 mg/kg/day PO in 3 divided doses (third-line)"
- Monitoring: Ferritin every 3 months (target less than 1000 μg/L); cardiac MRI T2* and liver iron quantification if available
- Evidence: Observational data suggest improved survival in transfusion-dependent lower-risk MDS patients receiving chelation vs. non-chelated historical controls [13]
8. Platelet and Neutrophil Support
- Platelet Transfusion: For bleeding or platelets less than 10 × 10⁹/L (prophylactic threshold)
- G-CSF (filgrastim): 300 μg SC 2-3x/week for recurrent infections in neutropenic patients; use cautiously (theoretical concern of promoting leukaemic transformation, though evidence limited)
- Antibiotic Prophylaxis: Consider in severe neutropenia (ANC less than 0.5) with recurrent infections
Higher-Risk MDS (IPSS-R Intermediate / High / Very High)
Goals: Prolong survival, delay AML transformation, achieve cytopenias control, bridge to transplant
1. Azacitidine (First-Line Standard) [7,18]
- Indication: Higher-risk MDS (IPSS-R Int/High/Very High); not transplant candidate or bridging to transplant
- Mechanism: Hypomethylating agent; DNA methyltransferase inhibitor; promotes differentiation and apoptosis of malignant clones
- Dose: 75 mg/m² SC daily days 1-7 every 28 days (7-day schedule); alternative 5-2-2 schedule (days 1-5, 8-9)
- Evidence: AZA-001 trial: Median OS 24.5 months (azacitidine) vs 15 months (conventional care regimens); 2-year survival 51% vs 26%; delayed AML transformation [7]
- Response:
- "Overall response rate: 40-50% (CR 10-20%, PR 10-15%, HI 15-20%)"
- "Median time to response: 3-6 cycles"
- "Median response duration: 12-18 months"
- Treatment Duration: Continue until progression, loss of response, or unacceptable toxicity; minimum 6 cycles to assess response
- Toxicity: Myelosuppression (grade 3-4 in 60-90%; nadir days 14-21; recover before next cycle), infection, injection site reactions, nausea, fatigue
- Monitoring: FBC weekly during cycle 1-2, then days 14-21 of subsequent cycles; delay/dose-reduce for prolonged myelosuppression
- Failure: Median survival after azacitidine failure is 4-6 months; consider clinical trial, best supportive care, or allo-HSCT if eligible
2. Decitabine (Alternative Hypomethylating Agent) [18]
- Indication: Higher-risk MDS; alternative to azacitidine (similar efficacy, no head-to-head superiority)
- Dose: 20 mg/m² IV over 1 hour daily × 5 days every 28 days
- Efficacy: Overall response 30-40%; CR 15-20%
- Toxicity: Similar myelosuppression profile to azacitidine
- Note: No survival advantage demonstrated over supportive care in European trials (unlike azacitidine); less commonly used in Europe; standard option in some regions
3. Allogeneic Haematopoietic Stem Cell Transplantation (Allo-HSCT) [8,14,18]
- Indication: Only potentially curative therapy for MDS; consider in IPSS-R Intermediate/High/Very High risk patients
- Candidacy:
- "Age: Typically less than 75 years (biological age and fitness more important than chronological age)"
- "Comorbidities: HCT-CI score to assess transplant risk"
- "Donor availability: Matched sibling, matched unrelated, haploidentical"
- Timing:
- "IPSS-R High/Very High: Early transplant preferred (after achieving disease control with azacitidine if high blast count)"
- "IPSS-R Intermediate: Controversial; balance transplant-related mortality vs. disease progression risk"
- "IPSS-R Low/Very Low: Generally not recommended unless high-risk features (e.g., TP53 mutation)"
- Pre-Transplant Therapy:
- May use azacitidine to reduce blast count and stabilize disease (2-6 cycles)
- Intensive chemotherapy (AML-like induction) if blasts > 10% and fit patient
- Conditioning Regimen:
- "Myeloablative conditioning (MAC): Younger, fit patients"
- "Reduced-intensity conditioning (RIC): Older patients, comorbidities; lower TRM but higher relapse risk"
- Outcomes:
- 3-year overall survival: 35-50% (varies by age, donor, disease risk, conditioning)
- 3-year relapse-free survival: 30-40%
- "Non-relapse mortality: 15-30% (depends on conditioning, donor match, age)"
- "TP53-mutated MDS: Very poor transplant outcomes (3-year OS 10-20%); relapse rates > 70% [12]"
4. Intensive Chemotherapy (Selected Cases) [18]
- Indication: MDS-IB2 (10-19% blasts), younger fit patients, bridge to allo-HSCT
- Regimen: AML-like induction (e.g., 7+3: cytarabine + anthracycline)
- Response: CR rates 40-60% (lower than de novo AML)
- Limitation: High relapse rate without consolidative allo-HSCT; not recommended as definitive therapy
- Role: Cytoreduction prior to transplant in high-blast-count MDS
5. Clinical Trials
- Encouraged for all higher-risk MDS patients, especially after azacitidine failure
- Investigational agents: IDH inhibitors (IDH1/2 mutated), FLT3 inhibitors, BCL-2 inhibitors (venetoclax combinations), magrolimab (CD47 inhibitor), checkpoint inhibitors
6. Best Supportive Care
- Transfusion support (RBC, platelets)
- Infection management (antibiotics, antifungals)
- Palliative care involvement for symptom management, advance care planning
Special Populations
TP53-Mutated MDS: [12,18]
- Very poor prognosis (median OS 9-12 months)
- Poor response to hypomethylating agents (response rate less than 20%)
- Poor allo-HSCT outcomes (high relapse, 3-year OS 10-20%)
- Management: Consider clinical trials (e.g., APR-246/eprenetapopt, TP53-targeted therapies), palliative approach in frail patients
Therapy-Related MDS (t-MDS):
- Generally higher-risk disease, complex karyotype, TP53 mutations
- Poor response to conventional therapy
- Allo-HSCT recommended in eligible patients
Elderly/Frail Patients:
- Careful assessment of fitness, comorbidities, performance status
- Lower-intensity hypomethylating agents well-tolerated in elderly
- Allo-HSCT with RIC feasible in selected patients up to age 75-80 years with good performance status
Treatment Algorithm Summary
MDS Diagnosis Confirmed
|
├─ Risk Stratify (IPSS-R, Molecular)
|
├─ IPSS-R Very Low / Low / Int (selected)
| └─ Symptomatic Anaemia?
| ├─ Yes → EPO less than 500? → ESA ± G-CSF
| | del(5q)? → Lenalidomide
| | RS/SF3B1? → Luspatercept (if ESA failed)
| | Transfusion-dependent → Chelation (if ferritin > 1000, life expectancy > 1y)
| └─ No → Watch and wait
|
└─ IPSS-R Intermediate / High / Very High
└─ Transplant Candidate?
├─ Yes → Azacitidine → Allo-HSCT (optimal timing per risk group)
└─ No → Azacitidine until progression → Clinical trial or BSC
8. Complications
Disease-Related Complications
| Complication | Incidence | Mechanism | Management |
|---|---|---|---|
| Transformation to AML | 30% cumulative (varies 5-80% by subtype) | Clonal evolution, acquisition of additional mutations | Intensive chemotherapy and/or allo-HSCT if eligible; otherwise BSC [9] |
| Severe infection | 20-40% | Neutropenia (ANC less than 0.5), neutrophil dysfunction | Broad-spectrum antibiotics; G-CSF support; prophylaxis in recurrent infections |
| Severe bleeding | 10-20% | Thrombocytopenia (less than 10-20), platelet dysfunction | Platelet transfusion; antifibrinolytics (tranexamic acid) for mucosal bleeding |
| Cardiac dysfunction (anaemia) | 15-25% | High-output state, chronic severe anaemia | Transfusion support to maintain Hb 80-100 g/L |
| Iron overload | 50-70% of transfusion-dependent | Transfusional iron accumulation (200-250 mg/unit) | Iron chelation when ferritin > 1000 μg/L [13] |
Treatment-Related Complications
Azacitidine/Decitabine: [7]
- Myelosuppression (60-90%): Prolonged cytopenias requiring transfusions, G-CSF
- Infection (30-50%): Neutropenic sepsis during nadir (days 14-21); empirical broad-spectrum antibiotics
- Gastrointestinal (30-40%): Nausea, diarrhoea, constipation; antiemetics
- Injection site reactions (50-70% with SC azacitidine): Erythema, bruising, pain; rotate sites, local measures
- Tumour lysis syndrome (less than 5%): In high-burden disease; hydration, allopurinol
Lenalidomide: [5]
- Myelosuppression (80%): Grade 3-4 neutropenia, thrombocytopenia; requires dose reductions/interruptions in 80%
- Thromboembolism (5-10%): DVT, PE; consider prophylactic anticoagulation in high-risk patients
- Rash (30-40%): Pruritic erythematous rash; antihistamines, topical steroids
- Diarrhoea (30%): Supportive management
- Fatigue (25-30%)
Luspatercept: [6]
- Hypertension (10-15%): Monitor BP; antihypertensive therapy as needed
- Bone pain (10-15%): Analgesics
- Fatigue (20-30%)
- Thrombosis (less than 5%): Similar to lenalidomide concern
Allo-HSCT: [8,14]
- Graft-versus-host disease (GVHD): Acute 30-50%, chronic 40-60%; immunosuppression required
- Infection: Bacterial, viral (CMV, EBV), fungal (aspergillus); prolonged immunosuppression
- Graft failure (5-10%): Primary or secondary; requires salvage strategies
- Relapse (30-50%): Higher in higher-risk disease, TP53-mutated MDS
- Organ toxicity: Hepatic veno-occlusive disease, renal insufficiency, pulmonary complications
- Secondary malignancies: Long-term risk 5-10%
Transfusion-Related:
- Iron overload: Cardiac (arrhythmias, cardiomyopathy), hepatic (cirrhosis), endocrine (diabetes, hypogonadism)
- Transfusion reactions: Allergic, febrile, haemolytic (rare)
- Alloimmunization: Development of antibodies to RBC/platelet antigens; difficulty finding compatible units
- Transfusion-transmitted infections: Very rare with modern screening
9. Prognosis
Overall Survival by IPSS-R
| IPSS-R Risk Category | Median Overall Survival | 5-Year OS | Median Time to 25% AML Transformation |
|---|---|---|---|
| Very Low | 8.8 years | 65% | Not reached (> 10 years) |
| Low | 5.3 years | 45% | 10.8 years |
| Intermediate | 3.0 years | 25% | 3.2 years |
| High | 1.6 years | 15% | 1.4 years |
| Very High | 0.8 years | 5% | 0.7 years |
[Data from Greenberg et al., IPSS-R cohort] [4]
Prognostic Factors
Favourable Prognostic Factors:
- Cytogenetics: Isolated del(5q), normal karyotype, del(20q), -Y
- Molecular: SF3B1 mutation (without high-risk co-mutations), isolated TET2/DNMT3A
- Clinical: Lower blast percentage (less than 5%), single-lineage cytopenia, transfusion-independence, younger age (less than 60 years)
- IPSS-R: Very Low or Low risk category
Unfavourable Prognostic Factors:
- Cytogenetics: Complex karyotype (≥3 abnormalities), monosomy 7/del(7q), inv(3)/t(3q), monosomy 5/del(5q) with other abnormalities
- Molecular: TP53 mutation (especially multi-hit), ASXL1, RUNX1, EZH2, multi-mutational burden (> 3 mutations) [10,12]
- Clinical: Higher blast percentage (> 10%), severe cytopenias (Hb less than 8, ANC less than 0.8, platelets less than 50), transfusion-dependence, older age (> 75 years)
- IPSS-R: High or Very High risk category
- Disease type: Therapy-related MDS (t-MDS)
Molecular Prognostic Subgroups
TP53-Mutated MDS: [12]
- Median OS: 9-12 months (mono-allelic), 8-10 months (multi-hit/biallelic)
- Response to azacitidine: less than 20%
- Allo-HSCT outcomes: 3-year OS 10-20%, relapse rate > 70%
- Represents 5-10% of all MDS, 20-30% of therapy-related MDS, 30-40% of MDS with complex karyotype
- Multi-hit TP53 (≥2 mutations, or mutation + loss of heterozygosity/del(17p)) worse than mono-allelic
SF3B1-Mutated MDS: [10]
- Median OS: 5-6 years
- Strong association with ring sideroblasts (> 90%)
- Favourable prognosis unless co-mutated with high-risk genes (TP53, RUNX1, ASXL1)
- Predicts response to luspatercept (60-70% transfusion-independence in SF3B1+ vs 30-40% SF3B1-) [6]
ASXL1, RUNX1, EZH2:
- Individually associated with shorter OS (median 1.5-2.5 years)
- Additive effect when multiple present
Impact of Treatment on Survival
Azacitidine in Higher-Risk MDS: [7]
- Median OS: 24.5 months (azacitidine) vs 15 months (conventional care)
- 2-year OS: 51% vs 26%
- Benefit greatest in IPSS Intermediate-2 and High risk (original IPSS classification)
Allogeneic HSCT: [8,14]
- 5-year OS: 30-50% (varies by age, risk category, donor type, conditioning)
- Curative potential: 30-40% long-term disease-free survival
- Best outcomes: IPSS-R Intermediate/High, age less than 65 years, matched sibling donor, TP53 wild-type
Lenalidomide in del(5q) MDS: [5]
- Median OS: 4-5 years in transfusion-dependent del(5q) MDS treated with lenalidomide
- Cytogenetic response associated with longer OS and lower AML transformation
Luspatercept in MDS-RS: [6]
- Reduction in transfusion burden associated with improved quality of life
- Overall survival data maturing
Quality of Life and Patient-Reported Outcomes
- Fatigue: Most common symptom affecting QoL; improvement with transfusion support, ESA, or luspatercept
- Transfusion burden: Major determinant of QoL; frequent hospital visits, time commitment, psychological impact
- Bleeding/infection symptoms: Anxiety, activity limitation
- Treatment toxicity: Azacitidine/lenalidomide myelosuppression may worsen short-term QoL before improvement
10. Evidence Base and Guidelines
Key Clinical Practice Guidelines
-
National Comprehensive Cancer Network (NCCN) Guidelines: Myelodysplastic Syndromes, Version 1.2025 — Comprehensive evidence-based recommendations for diagnosis, risk stratification, and treatment of MDS across all risk categories. Regularly updated with emerging data.
-
European LeukemiaNet (ELN) Recommendations for Diagnosis and Management of MDS (2022) — International expert consensus on diagnostic criteria, prognostic assessment, and therapeutic algorithms. PMID: 34773327 [18]
-
British Society for Haematology (BSH) Guidelines: Diagnosis and Management of Adult Myelodysplastic Syndromes (2014, updated 2023) — UK-specific guidance on investigation, treatment, and supportive care strategies.
-
WHO Classification of Haematolymphoid Tumours (5th Edition, 2022) — Definitive classification system integrating morphology, cytogenetics, and molecular features. PMID: 35931190 [3]
-
International Working Group (IWG) Response Criteria for MDS (2023 Update) — Standardized response definitions for clinical trials and practice.
Landmark Clinical Trials
1. IPSS-R Development and Validation (Greenberg et al., Blood 2012) [4]
- Design: International cohort study, 7,012 MDS patients
- Objective: Develop refined prognostic scoring system
- Findings: IPSS-R incorporating 5-category cytogenetics, depth of cytopenias, and blast % provides superior prognostic discrimination compared to original IPSS
- Impact: Gold standard for MDS risk stratification; guides treatment decisions globally
- PMID: 22740453
2. AZA-001 Trial (Fenaux et al., Lancet Oncol 2009; Fenaux et al., J Clin Oncol 2010) [7]
- Design: Phase III randomized controlled trial, 358 higher-risk MDS patients (IPSS Intermediate-2/High)
- Arms: Azacitidine 75 mg/m² SC days 1-7 q28d vs conventional care regimens (best supportive care, low-dose cytarabine, or intensive chemotherapy)
- Primary Endpoint: Overall survival
- Key Findings:
- "Median OS: 24.5 months (azacitidine) vs 15.0 months (conventional care); HR 0.58, pless than 0.0001"
- 2-year OS: 51% vs 26%
- "Time to AML transformation: 17.8 months vs 11.5 months"
- "Response rate: 49% (CR 17%, PR 12%, HI 20%)"
- Impact: Established azacitidine as standard of care for higher-risk MDS; first agent to demonstrate survival benefit
- PMID: 19230772 (primary), 20516417 (survival update)
3. MDS-003 and MDS-004 Lenalidomide Trials (List et al., NEJM 2006) [5]
- Design: Two phase II trials in transfusion-dependent MDS with del(5q)
- Treatment: Lenalidomide 10 mg or 5 mg daily
- Key Findings:
- "Transfusion independence: 67% (10 mg), 56% (5 mg)"
- "Cytogenetic complete response: 45%"
- "Median duration of transfusion independence: 2+ years"
- "Median time to response: 4-5 weeks"
- Impact: Established lenalidomide as standard therapy for del(5q) MDS; FDA approval 2005
- PMID: 17021321
4. MEDALIST Trial (Fenaux et al., NEJM 2020) [6]
- Design: Phase III randomized, double-blind, placebo-controlled trial, 229 patients with MDS-RS, IPSS-R Very Low/Low/Intermediate, transfusion-dependent, ESA-failure or high EPO
- Arms: Luspatercept (1.0-1.75 mg/kg SC q3weeks) vs placebo
- Primary Endpoint: Transfusion independence ≥8 weeks during weeks 1-24
- Key Findings:
- "Transfusion independence ≥8 weeks: 38% (luspatercept) vs 13% (placebo); pless than 0.001"
- "Transfusion independence ≥12 weeks: 28% vs 8%"
- ≥50% reduction in transfusion burden: 53% vs 19%
- "SF3B1 mutation: Strong predictor of response"
- Impact: FDA/EMA approval for luspatercept in MDS-RS; new standard for transfusion-dependent lower-risk MDS-RS after ESA failure
- PMID: 31914241
5. Decitabine Phase III Trials (Kantarjian et al., JCO 2006; Steensma et al., JCO 2009)
- Design: Phase III trials of decitabine vs supportive care in higher-risk MDS
- Findings: Improved response rates (CR 15-17%), delayed AML transformation, improved QoL; no OS benefit in European trial (unlike azacitidine)
- Impact: Decitabine approved for MDS; azacitidine preferred in Europe due to survival data
- PMID: 16314617, 19075274
6. Transplant Timing Studies (Cutler et al., Blood 2004; Koreth et al., JCO 2013) [14]
- Design: Decision analysis and Markov modeling to determine optimal timing of allo-HSCT
- Key Findings:
- "IPSS High/Intermediate-2: Early transplant maximizes survival"
- "IPSS Intermediate-1: Delayed transplant until progression may maximize quality-adjusted life years"
- Age, comorbidities, donor availability critical variables
- Impact: Informs transplant decision-making; no one-size-fits-all approach
- PMID: 15054039, 23109696
7. TP53-Mutated MDS Analyses (Bernard et al., JCO 2020; Haase et al., Leukemia 2019) [12]
- Design: Large cohort studies analyzing outcomes in TP53-mutated MDS
- Key Findings:
- "TP53 mutations (mono-allelic): Median OS 15-18 months"
- "TP53 multi-hit (biallelic): Median OS 9-12 months"
- "Response to azacitidine: less than 20%"
- "Post-allo-HSCT relapse: 70-80% at 3 years"
- Poor outcomes regardless of therapy
- Impact: Recognize TP53-mutated MDS as ultra-high-risk subgroup; prioritize clinical trials
- PMID: 31682526, 30692665
8. Iron Chelation in MDS (Leitch et al., Leuk Res 2011; Lyons et al., Br J Haematol 2017) [13]
- Design: Retrospective observational studies of chelation vs non-chelation in transfusion-dependent lower-risk MDS
- Findings: Improved OS in chelated vs non-chelated patients in observational cohorts; confounding by indication (chelated patients healthier); no randomized data
- Recommendation: Guideline consensus supports chelation in transfusion-dependent lower-risk MDS with life expectancy > 1 year
- PMID: 21324525, 28832962
Emerging Evidence and Future Directions
Molecular-Integrated Prognostic Systems:
- IPSS-M (Molecular IPSS): Incorporates 31 recurrent mutations; improved prognostic discrimination; validation ongoing
- Personalized risk stratification using comprehensive molecular profiling
Targeted Therapies in Development:
- IDH1/2 inhibitors (ivosidenib, enasidenib): In IDH-mutated MDS
- FLT3 inhibitors: In FLT3-mutated MDS/AML
- BCL-2 inhibitor (venetoclax): Combination with azacitidine (ongoing trials)
- TP53-targeted therapies (APR-246/eprenetapopt): In TP53-mutated MDS (trials ongoing)
- Magrolimab (anti-CD47): Macrophage checkpoint inhibitor (phase III trials)
- Sabatolimab (anti-TIM-3): Checkpoint inhibitor (phase III trials)
Transplant Innovations:
- Haploidentical transplant with post-transplant cyclophosphamide: Expanding donor pool
- Reduced-intensity conditioning optimization
- Post-transplant maintenance (azacitidine, lenalidomide) to reduce relapse
11. Patient Education and Shared Decision-Making
What Are Myelodysplastic Syndromes?
Myelodysplastic syndromes (MDS) are a group of blood disorders where your bone marrow—the spongy tissue inside your bones that makes blood cells—doesn't work properly. In MDS, the bone marrow produces abnormal blood cells that don't mature normally. As a result, you end up with low numbers of healthy blood cells in your bloodstream.
There are three main types of blood cells affected:
- Red blood cells carry oxygen around your body. Low red cells (anaemia) causes tiredness, shortness of breath, and weakness.
- White blood cells fight infections. Low white cells (neutropenia) increases your risk of infections.
- Platelets help your blood clot. Low platelets (thrombocytopenia) causes bruising and bleeding.
MDS mainly affects older adults, with most people diagnosed in their 70s.
What Causes MDS?
In most cases, we don't know exactly why someone develops MDS. It happens more often as people get older. Sometimes MDS can develop after chemotherapy or radiotherapy for another cancer (called therapy-related MDS). Rarely, it can be linked to exposure to certain chemicals like benzene.
How Is MDS Diagnosed?
Your doctor will need to:
- Take blood tests to measure your blood cell counts and examine blood cells under a microscope
- Perform a bone marrow biopsy, where a small sample of bone marrow is removed (usually from your hip bone) using a needle. This is done under local anaesthetic. The sample is examined to look for abnormal cells and genetic changes.
- Order genetic tests on your bone marrow to help determine your prognosis and guide treatment.
What Is My Prognosis?
MDS varies widely from person to person. Your doctor will use a scoring system called IPSS-R that looks at genetic abnormalities, blast cell percentage, and blood counts to categorize your MDS as "lower-risk" or "higher-risk."
- Lower-risk MDS: Can remain stable for many years; median survival 5-9 years. Focus is on managing symptoms and improving quality of life.
- Higher-risk MDS: More likely to progress to acute leukaemia; median survival 1-3 years. Treatment aims to slow disease progression and prolong survival.
About 30% of people with MDS will eventually develop acute myeloid leukaemia (AML), though this varies greatly depending on your specific MDS type.
What Are My Treatment Options?
Treatment depends on whether you have lower-risk or higher-risk MDS:
For Lower-Risk MDS:
- Watchful waiting: If you have no or mild symptoms, you may not need treatment right away. Your doctor will monitor your blood counts regularly.
- Blood transfusions: Red blood cell transfusions for anaemia when you become symptomatic. Platelet transfusions if you have severe bleeding.
- Erythropoietin injections: A hormone that stimulates red blood cell production, reducing the need for transfusions in some people.
- Lenalidomide tablets: If you have a specific genetic abnormality called del(5q), this medication can reduce or eliminate the need for transfusions in about 2 out of 3 people.
- Luspatercept injections: If you have ring sideroblasts (a specific type of MDS), this medication can reduce transfusion needs.
- Iron chelation therapy: If you're receiving regular transfusions, iron can build up in your body and damage organs. Iron chelation removes excess iron.
For Higher-Risk MDS:
- Azacitidine injections: The main medication for higher-risk MDS, given as injections under the skin for 7 days every 4 weeks. It helps control the disease and prolongs survival.
- Stem cell transplant: The only treatment that can potentially cure MDS. It involves high-dose chemotherapy followed by infusion of healthy stem cells from a donor. This is a major procedure with significant risks and is only suitable for fitter, younger patients (usually under 75 years).
What Are the Side Effects of Treatment?
- Transfusions: Generally safe; mild reactions (fever, chills) occasionally occur. Long-term transfusions cause iron buildup.
- Azacitidine: Commonly causes low blood counts (increasing infection and bleeding risk), injection site reactions, nausea, and fatigue. Your blood counts will be monitored closely.
- Lenalidomide: Can cause severe drops in blood counts requiring dose adjustments, and a small increased risk of blood clots.
- Stem cell transplant: Major risks include graft-versus-host disease (where the donor cells attack your body), severe infections, and organ damage. Your transplant team will discuss these in detail.
Living with MDS
- Infection prevention: Wash hands frequently, avoid crowds during flu season, report fevers (> 38°C) promptly.
- Bleeding precautions: Avoid contact sports, use soft toothbrush, avoid NSAIDs (ibuprofen) which affect platelets.
- Fatigue management: Pace activities, prioritize important tasks, consider occupational therapy referral.
- Emotional support: MDS diagnosis can be overwhelming. Consider support groups, counselling, or connecting with MDS advocacy organizations.
- Advance care planning: Discuss your wishes for future care with your doctor and loved ones.
Questions to Ask Your Doctor
- What is my IPSS-R risk category?
- What are my treatment options and their goals (symptom control vs prolonging survival)?
- Am I a candidate for stem cell transplant?
- How often will I need transfusions?
- What are the signs of infection or bleeding I should watch for?
- Are there clinical trials available for me?
- How will MDS affect my daily life and activities?
12. References
-
Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts: Myelodysplastic Syndromes. National Cancer Institute. Available at: https://seer.cancer.gov/statfacts/html/mulmy.html [Accessed 2025]
-
Cazzola M. Myelodysplastic syndromes. N Engl J Med. 2020;383(14):1358-1374. PMID: 33027576. DOI: 10.1056/NEJMra1904794
-
Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200-1228. PMID: 35931190. DOI: 10.1182/blood.2022015850
-
Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood. 2012;120(12):2454-2465. PMID: 22740453. DOI: 10.1182/blood-2012-03-420489
-
List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456-1465. PMID: 17021321. DOI: 10.1056/NEJMoa061292
-
Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. 2020;382(2):140-151. PMID: 31914241. DOI: 10.1056/NEJMoa1908892
-
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223-232. PMID: 19230772. DOI: 10.1016/S1470-2045(09)70003-8
-
Della Porta MG, Alessandrino EP, Bacigalupo A, et al. Predictive factors for the outcome of allogeneic transplantation in patients with MDS stratified according to the revised IPSS-R. Blood. 2014;123(15):2333-2342. PMID: 24558199. DOI: 10.1182/blood-2013-12-542720
-
Malcovati L, Hellström-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943-2964. PMID: 23980065. DOI: 10.1182/blood-2013-03-492884
-
Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241-247. PMID: 24220272. DOI: 10.1038/leu.2013.336
-
Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood. 2008;111(2):574-582. PMID: 17940203. DOI: 10.1182/blood-2007-06-096370
-
Bernard E, Nannya Y, Hasserjian RP, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26(10):1549-1556. PMID: 32929281. DOI: 10.1038/s41591-020-1008-z
-
Leitch HA, Goodman TA, Wong KK, et al. Improved survival in patients with myelodysplastic syndrome receiving iron chelation therapy. Clin Leuk. 2008;2(3):205-211. PMID: 24533269. DOI: 10.3816/CLK.2008.n.023
-
Koreth J, Pidala J, Perez WS, et al. Role of reduced-intensity conditioning allogeneic hematopoietic stem-cell transplantation in older patients with de novo myelodysplastic syndromes: an international collaborative decision analysis. J Clin Oncol. 2013;31(21):2662-2670. PMID: 23797000. DOI: 10.1200/JCO.2012.46.8652
-
Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001-2004, using data from the NAACCR and SEER programs. Blood. 2008;112(1):45-52. PMID: 18443215. DOI: 10.1182/blood-2008-01-134858
-
Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. PMID: 25426837. DOI: 10.1056/NEJMoa1408617
-
Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496-2506. PMID: 21714648. DOI: 10.1056/NEJMoa1013343
-
Platzbecker U, Fenaux P, Adès L, et al. Proposals for revised IWG 2023 response criteria for higher risk MDS. Blood. 2023;141(21):2047-2061. PMID: 36580613. DOI: 10.1182/blood.2022018603
-
Garcia-Manero G, Sasaki K, Montalban-Bravo G, et al. A phase II study of TP53 modulator APR-246 in TP53-mutant MDS and oligoblastic AML. Blood. 2021;138(Suppl 1):3401. DOI: 10.1182/blood-2021-147856
-
Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9-16. PMID: 25931582. DOI: 10.1182/blood-2015-03-631747
-
Sekeres MA, Guyatt G, Abel G, et al. American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020;4(15):3528-3549. PMID: 32759088. DOI: 10.1182/bloodadvances.2020001920
-
Komrokji RS, Padron E, Ebert BL, et al. Therapy-related myeloid neoplasms: when genetics and therapy collide. Blood Rev. 2019;37:100583. PMID: 31208786. DOI: 10.1016/j.blre.2019.100583
13. Examination Preparation
Viva Voce Key Points
Opening Statement: "Myelodysplastic syndromes are clonal haematopoietic stem cell disorders characterized by ineffective haematopoiesis resulting in peripheral cytopenias despite normocellular or hypercellular bone marrow, morphological dysplasia in one or more myeloid lineages, and risk of transformation to acute myeloid leukaemia. The diagnosis requires bone marrow examination, and risk stratification using the IPSS-R is essential to guide treatment."
Essential Facts for Viva:
- Definition: Clonal stem cell disorders; dysplasia ≥10% in ≥1 lineage; cytopenias; AML risk
- Epidemiology: Median age 70-75 years; incidence 4-5 per 100,000, rising to > 30 per 100,000 in elderly
- Diagnosis: Mandatory bone marrow examination with morphology, cytogenetics, molecular studies; exclude B12/folate deficiency
- Classification: WHO 2022: MDS-LB, MDS-RS, MDS-IB1, MDS-IB2, MDS-del(5q), hypoplastic/fibrotic variants
- IPSS-R: Five risk groups (Very Low to Very High) based on cytogenetics, blasts, Hb, platelets, ANC
- Lower-Risk Treatment: Supportive care, ESAs (if EPO less than 500), lenalidomide (del(5q)), luspatercept (RS), transfusion support, iron chelation
- Higher-Risk Treatment: Azacitidine 75 mg/m² SC days 1-7 q28d (standard first-line), allo-HSCT (only curative option)
- Molecular Mutations: SF3B1 (favourable, RS, luspatercept-responsive), TP53 (very poor, median OS 9-12 months), ASXL1/RUNX1/EZH2 (poor)
- AML Transformation: 30% cumulative incidence; varies by subtype (5% del(5q) to 80% MDS-IB2)
- Prognosis: IPSS-R Very Low (8.8 years) to Very High (0.8 years median OS)
High-Yield Topics for Written Examinations
MCQ/SBA Pearls:
- Diagnostic Criteria: Dysplasia ≥10% in ≥1 lineage is required for MDS diagnosis
- IPSS-R: Incorporates 5-category cytogenetics (Very Good to Very Poor), blasts, and 3 cytopenias
- del(5q): Lenalidomide first-line; 67% transfusion independence
- SF3B1 Mutation: Associated with ring sideroblasts (> 90%); predicts luspatercept response
- TP53 Mutation: Very poor prognosis; median OS less than 12 months; poor azacitidine and transplant response
- Azacitidine: Only agent proven to prolong survival in higher-risk MDS (AZA-001 trial: 24.5 vs 15 months)
- EPO Level: Best predictor of ESA response; less than 500 mU/mL = 60-70% response
- Iron Chelation: Initiate when ferritin > 1000 μg/L, transfusion-dependent, life expectancy > 1 year
- Allo-HSCT: Only curative therapy; consider in IPSS-R Intermediate/High/Very High
- Exclude B12/Folate: Mandatory before diagnosing MDS (cause dysplasia and macrocytosis)
Common MRCP/FRACP Examination Mistakes
- ❌ Diagnosing MDS from peripheral blood alone → Bone marrow examination is mandatory
- ❌ Not checking B12/folate before diagnosing MDS → Always exclude nutritional causes
- ❌ Not calculating IPSS-R → Essential for treatment decisions
- ❌ Treating higher-risk MDS with only supportive care → Requires disease-modifying therapy (azacitidine or transplant)
- ❌ Missing del(5q) and not offering lenalidomide → High response rates, must check cytogenetics
- ❌ Giving ESA to patients with EPO > 500 mU/mL → Very low response rate (less than 10%)
- ❌ Not considering allo-HSCT in fit higher-risk patients → Only curative option
- ❌ Confusing MDS with CMML → CMML has monocytosis ≥1 × 10⁹/L (MDS/MPN overlap)
- ❌ Not initiating iron chelation in chronically transfused lower-risk MDS → Prevents cardiac/hepatic iron toxicity
Clinical Case Scenarios
Scenario 1: Lower-Risk MDS with del(5q)
- 72-year-old woman, macrocytic anaemia (Hb 85 g/L, MCV 108 fL), B12/folate normal
- Bone marrow: 10% erythroid dysplasia, 2% blasts, cytogenetics shows isolated del(5q)
- IPSS-R: Low risk
- Management: Lenalidomide 10 mg daily days 1-21; monitor for myelosuppression; 67% chance of transfusion independence
Scenario 2: Higher-Risk MDS
- 68-year-old man, pancytopenia (Hb 75 g/L, WBC 2.0, platelets 35), bone marrow 12% blasts, complex karyotype
- IPSS-R: Very High risk
- Management: Azacitidine 75 mg/m² SC days 1-7 q28d; assess allo-HSCT candidacy; median OS 24.5 months with azacitidine vs 15 months supportive care
Scenario 3: MDS-RS, ESA Failure
- 75-year-old woman, transfusion-dependent (4 units/month), bone marrow shows 25% ring sideroblasts, SF3B1 mutation, failed prior ESA
- IPSS-R: Low risk
- Management: Luspatercept 1.0 mg/kg SC q3weeks; 38% achieve transfusion independence ≥8 weeks (53% with SF3B1 mutation)
Last Updated: 2026-01-09