Myasthenia Gravis
Clinically, MG presents with ptosis and diplopia in 85% of cases at onset, progressing to generalised weakness in 50-80% within 2 years. Bulbar symptoms (dysarthria, dysphagia) and proximal limb weakness are...
What matters first
Clinically, MG presents with ptosis and diplopia in 85% of cases at onset, progressing to generalised weakness in 50-80% within 2 years. Bulbar symptoms (dysarthria, dysphagia) and proximal limb weakness are...
Respiratory muscle weakness (MG crisis)
9 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Respiratory muscle weakness (MG crisis)
- Bulbar symptoms (dysphagia, dysarthria)
- Rapid deterioration
- FVC less than 1L or declining
Linked comparisons
Differentials and adjacent topics worth opening next.
- Lambert-Eaton Myasthenic Syndrome
- Botulism
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Myasthenia Gravis
1. Clinical Overview
Summary
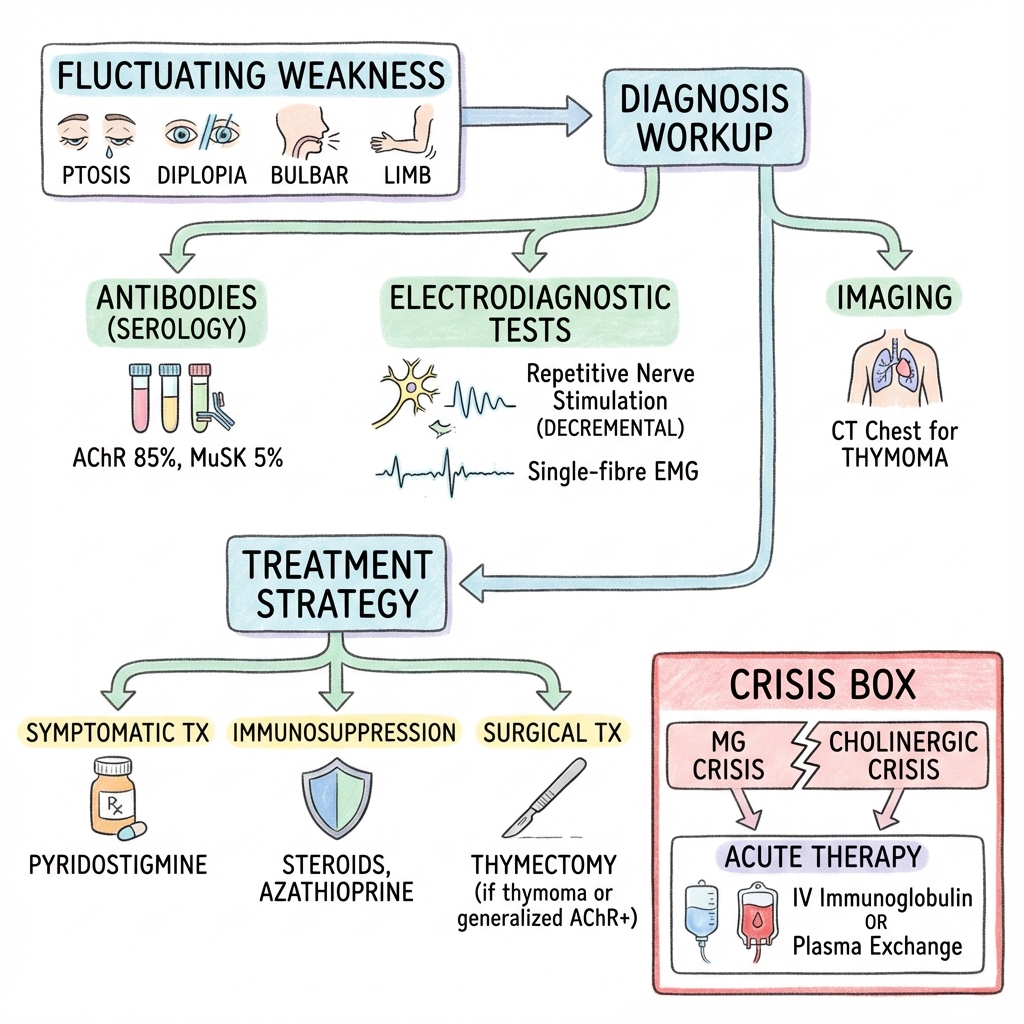
Myasthenia gravis (MG) is an autoimmune disorder of the neuromuscular junction (NMJ) characterised by fluctuating fatigable weakness of skeletal muscles that worsens with activity and improves with rest. The disease is mediated by antibodies directed against components of the postsynaptic membrane, most commonly the acetylcholine receptor (AChR), present in 80-85% of patients. [1,2] Alternative antibody targets include muscle-specific kinase (MuSK) in 5-8% and lipoprotein-related protein 4 (LRP4) in less than 1%, with approximately 10% of patients remaining seronegative for known antigens. [2,3]
Clinically, MG presents with ptosis and diplopia in 85% of cases at onset, progressing to generalised weakness in 50-80% within 2 years. [4] Bulbar symptoms (dysarthria, dysphagia) and proximal limb weakness are characteristic. The hallmark fatigability—weakness worsening with repetitive activity and improving after rest—distinguishes MG from other neuromuscular disorders. Myasthenic crisis, defined as respiratory failure requiring mechanical ventilation, occurs in 15-20% of patients and represents a medical emergency with mortality under 5% in modern intensive care. [5]
Diagnosis is established through antibody testing (AChR, MuSK, LRP4), electrophysiology demonstrating decremental response on repetitive nerve stimulation (RNS) or increased jitter on single-fibre EMG, and clinical response to acetylcholinesterase inhibitors. CT or MRI of the thorax is mandatory in all patients to detect thymoma, present in 10-20% of AChR-positive cases. [2,6]
Treatment is multimodal: symptomatic management with pyridostigmine (60-120mg four times daily), immunosuppression with corticosteroids and steroid-sparing agents (azathioprine, mycophenolate), and thymectomy for thymomatous or non-thymomatous generalised AChR-positive MG under age 65, supported by the landmark MGTX trial. [7] MuSK-positive MG shows poor response to pyridostigmine and acetylcholinesterase inhibitors, with rituximab emerging as particularly effective. [8,9] Acute exacerbations and crisis require plasmapheresis or intravenous immunoglobulin (IVIG). [10]
With modern treatment, prognosis is excellent with near-normal life expectancy. Mortality has decreased from 30-40% historically to less than 5% currently, primarily driven by advances in intensive care management of myasthenic crisis. [5] Medication-free remission occurs in 20-30% of patients. [4]
Key Facts
- Definition: Autoimmune NMJ disorder with antibodies against AChR (80-85%), MuSK (5-8%), or LRP4 (less than 1%)
- Incidence: 8-30 per million per year (increasing with improved diagnosis and ageing populations) [11]
- Prevalence: 150-250 per million (regional variation) [11]
- Peak Demographics: Bimodal distribution—women 20-40 years (early-onset), men 60-80 years (late-onset) [4,11]
- Pathognomonic Feature: Fluctuating fatigable weakness improving with rest
- Gold Standard Investigation: AChR antibodies (85% sensitive in generalised MG, 50% in ocular MG)
- Thymoma Association: 10-20% of AChR-positive MG; mandatory imaging in all cases [6]
- First-line Treatment: Pyridostigmine (symptomatic) + immunosuppression (corticosteroids, steroid-sparing agents)
- Crisis Management: ICU care, plasmapheresis or IVIG, mechanical ventilation if FVC less than 15mL/kg
- Prognosis: Mortality less than 5% with modern treatment; 20-30% achieve remission [4,5]
Clinical Pearls
Diagnostic Pearl: The ice pack test—applying ice to ptotic eyelid for 2 minutes—improves ptosis by ≥2mm in 80-90% of ocular MG, providing immediate bedside confirmation. Cooling slows acetylcholinesterase degradation, increasing ACh availability. [12]
Antibody Pearl: If AChR antibodies are negative in clinically suspected generalised MG, test for MuSK antibodies (5-8% of cases) and LRP4 (1-3%). MuSK-positive patients have distinctive phenotype: female predominance, bulbar and neck weakness, muscle atrophy, poor pyridostigmine response, and excellent rituximab response. [3,9]
Steroid Pearl: When initiating corticosteroids in MG, start LOW (10mg prednisolone) and increase gradually (5mg every 3-5 days). High-dose initiation (e.g., 60mg) can precipitate acute worsening in 30-50% of patients within 7-10 days, potentially requiring ICU admission. [1,13]
Crisis Pearl: Distinguish myasthenic crisis (under-treatment: weakness, tachycardia, sweating, dilated pupils) from cholinergic crisis (over-treatment with acetylcholinesterase inhibitors: weakness plus excessive secretions, miosis, bradycardia, diarrhoea). Temporarily stopping pyridostigmine aids diagnosis—improvement suggests cholinergic crisis. [13]
Drug Safety Pearl: Aminoglycosides (gentamicin, tobramycin), fluoroquinolones, and macrolides can precipitate myasthenic crisis by impairing neuromuscular transmission. D-penicillamine can induce MG de novo. Beta-blockers and calcium channel blockers should be avoided. Always screen medications before prescribing in MG. [14]
Why This Matters Clinically
MG is a treatable cause of progressive weakness that, if unrecognised, can lead to life-threatening respiratory failure. Early diagnosis and appropriate immunosuppression prevent progression to crisis, maintain quality of life, and in many cases achieve medication-free remission. Understanding the approach to myasthenic crisis—including rapid airway assessment, FVC monitoring, and acute immunomodulation—is critical for acute medicine, neurology, and intensive care practice. Recognising MG as a potential presenting feature of thymoma (10-20% of cases) ensures timely imaging and oncological management.
2. Epidemiology
Incidence and Prevalence
The epidemiology of MG has evolved significantly over recent decades, with increasing incidence and prevalence attributed to improved diagnostic techniques (antibody testing, electrophysiology), greater awareness, and population ageing. [11]
| Measure | Rate | Notes |
|---|---|---|
| Incidence | 8-30 per million per year | Wide geographic variation; higher in European and North American populations [11] |
| Prevalence | 150-250 per million | Increasing steadily; Japan reports 200 per million, Norway 250 per million [11] |
| Ocular MG | 15-20% remain purely ocular | 50-80% of ocular cases generalise within 2 years [4] |
| Myasthenic Crisis | 15-20% lifetime incidence | Usually occurs within first 2-3 years of disease [5] |
| Mortality | less than 5% (modern era) | Reduced from 30-40% pre-ICU era; crisis-related deaths driven by age and comorbidities [5] |
Demographics
Age Distribution
MG demonstrates a characteristic bimodal age distribution:
-
Early-Onset MG (EOMG): Peak at 20-40 years
- "Predominantly female (F:M = 3:1)"
- Associated with thymic hyperplasia (60-70%)
- Often AChR-positive with good treatment response
- Higher spontaneous remission rates (30-40%) [4,11]
-
Late-Onset MG (LOMG): Peak at 60-80 years
- "Predominantly male (M:F = 1.5-2:1)"
- Associated with thymic atrophy or normal thymus
- More severe disease course
- Lower remission rates
- Higher comorbidity burden [4,11]
Sex Distribution
- Overall F:M ratio approximately 3:2
- Female predominance in early-onset disease (F:M = 3:1)
- Male predominance in late-onset disease (M:F = 1:1.5-2) [11]
- MuSK-positive MG shows strong female predominance (F:M = 9:1) [3]
Ethnic Variation
- Higher prevalence in Asian populations for ocular MG
- MuSK antibodies more common in Mediterranean and African populations
- Genetic associations: HLA-B8, DR3, DQ2 in AChR-positive European populations; HLA-DR14, DQ5 in MuSK-positive patients [15]
Classification by Antibody Subtype
| Subtype | Frequency | Key Features | Treatment Response |
|---|---|---|---|
| AChR-positive | 80-85% | Classic generalised or ocular MG; thymic pathology common | Good response to all treatments including pyridostigmine, steroids, thymectomy |
| MuSK-positive | 5-8% | Female predominance; bulbar/neck/respiratory weakness; muscle atrophy; minimal ocular involvement | Poor pyridostigmine response; excellent rituximab response; no benefit from thymectomy [3,9] |
| LRP4-positive | 1-3% | Milder phenotype; overlaps with AChR clinical features | Similar to AChR-positive disease |
| Seronegative | 5-10% | May have low-affinity AChR antibodies undetectable by standard assays or antibodies to other unknown antigens | Variable treatment response; often similar to AChR-positive |
Thymic Pathology Distribution
| Thymic Pathology | Frequency | Age Association | Clinical Significance |
|---|---|---|---|
| Thymic hyperplasia | 60-70% | EOMG (less than 40 years) | Responds well to thymectomy |
| Thymoma | 10-20% | LOMG (peak 40-60) | Mandatory thymectomy; potential malignancy (WHO classification) |
| Normal/atrophic | 20-30% | LOMG (> 60 years) | Thymectomy benefit less clear in elderly |
3. Aetiology and Pathophysiology
Aetiology
MG is a prototypical antibody-mediated autoimmune disease. The loss of immune tolerance to NMJ proteins is incompletely understood but involves thymic dysfunction, genetic susceptibility, and environmental triggers.
Genetic Factors
- HLA associations: HLA-B8, DR3, DQ2 (AChR-positive Europeans); HLA-DR14, DQ5 (MuSK-positive) [15]
- Non-HLA genes: PTPN22, CTLA4, TNFRSF11A (immune regulation)
- Family history increases risk 1000-fold compared to general population (though absolute risk remains low)
Thymic Abnormalities
- Thymic hyperplasia (60-70%): Germinal centre formation with B cells producing AChR antibodies
- Thymoma (10-20%): Neoplastic thymic epithelial cells express AChR-like proteins, triggering autoimmunity; 30-50% of thymoma patients develop MG [6]
- Thymus expresses AChR and is site of T-cell education; abnormalities drive autoreactive T-cell and B-cell activation
Triggers and Associations
- Infections: Molecular mimicry hypothesised (no definitive pathogen identified)
- Medications: D-penicillamine (induces MG in 1-7% of users; usually resolves after cessation) [14]
- Autoimmune associations: Thyroid disease (5-10%), SLE, rheumatoid arthritis, pernicious anaemia
- Pregnancy: Exacerbations in 30-40% (particularly postpartum)
Molecular Pathophysiology
The NMJ comprises the presynaptic motor nerve terminal, synaptic cleft, and postsynaptic muscle endplate membrane with densely packed AChRs.
Normal Neuromuscular Transmission
- Action potential reaches presynaptic terminal → voltage-gated calcium channels open
- Calcium influx triggers acetylcholine (ACh) vesicle fusion and release into synaptic cleft
- ACh binds to AChR (nicotinic pentamer: 2α, β, δ, ε subunits)
- AChR channel opens → sodium influx → endplate potential
- If threshold reached → muscle fibre action potential → contraction
- ACh degraded by acetylcholinesterase (AChE) in synaptic cleft
Safety Factor: Normally, ACh release far exceeds threshold (safety factor 4-10×), ensuring reliable transmission even with reduced receptor availability.
Pathophysiology in AChR-Positive MG
-
Antibody Production: B cells (often originating from thymus) produce IgG1 and IgG3 antibodies against AChR α-subunit (main immunogenic region)
-
Receptor Destruction: Three mechanisms:
- Complement-mediated lysis: Antibody binding activates complement cascade (C3, C5b-9 membrane attack complex) → postsynaptic membrane damage and simplification
- Accelerated receptor internalisation: Antibody cross-linking causes receptor endocytosis and degradation
- Functional blockade: Antibodies sterically block ACh binding (less common)
-
Reduced Receptor Density: Normal AChR density 10,000 per μm²; in MG reduced to less than 30% of normal
-
Impaired Transmission:
- At rest: Sufficient ACh released to reach threshold despite reduced receptors
- With repetitive activity: ACh depletion occurs faster; safety factor lost → transmission failure
- Results in fatigable weakness
Pathophysiology in MuSK-Positive MG
MuSK (muscle-specific kinase) is essential for AChR clustering at the NMJ via agrin-LRP4-MuSK signalling pathway.
- Antibody Mechanism: IgG4 antibodies against MuSK (non-complement fixing)
- Disrupted AChR Clustering: MuSK inhibition prevents AChR aggregation at postsynaptic membrane
- Neuromuscular Junction Dysfunction: Reduced AChR density and abnormal NMJ architecture
- No Complement Activation: Explains different clinical phenotype and treatment response compared to AChR-MG [3,9]
Pathophysiology in LRP4-Positive MG
LRP4 (lipoprotein receptor-related protein 4) acts as agrin receptor, initiating MuSK signalling. LRP4 antibodies disrupt agrin-LRP4-MuSK pathway, impairing AChR clustering. Clinical phenotype resembles AChR-positive MG. [3]
Why Ocular Muscles Are Particularly Affected
Extraocular muscles are disproportionately affected in MG for several reasons:
- High firing rates: Continuous activity maintaining gaze (high ACh demand)
- Small motor units: Fewer muscle fibres per motor neuron → less safety margin
- Different AChR subunit expression: Fetal γ-subunit (vs. adult ε-subunit) may have different immunogenicity
- Twitch fibre properties: Predominantly fast-twitch fibres with higher metabolic demand [4]
4. Clinical Presentation
Symptoms
Ocular (85% at presentation, 90% eventually)
- Ptosis: Unilateral or bilateral; fatigable (worsens with sustained upgaze); variable (fluctuates during day)
- Diplopia: Binocular; any direction; variable and fatigable
- Complex ophthalmoplegia: No pattern following single nerve distribution (distinguishes from cranial nerve palsy)
Bulbar (60% in generalised MG)
- Dysarthria: Nasal speech; voice fades with prolonged speaking ("myasthenic snarl" when attempting to smile)
- Dysphagia: Difficulty with liquids > solids (palatal weakness); nasal regurgitation
- Fatigable chewing: Jaw fatigue when eating; may need to support jaw with hand
- Facial weakness: Expressionless facies; weak eye closure
Limb (60% in generalised MG)
- Proximal > distal weakness: Shoulder abduction, hip flexion most affected
- Difficulty with overhead tasks: Combing hair, reaching high shelves
- Difficulty rising from chair: Without using arms
- Climbing stairs: Difficulty with repetitive leg movements
Neck
- Neck flexor weakness: "Head drop"—difficulty holding head upright, particularly after prolonged posture
- Neck extensor involvement: Less common than flexors
Respiratory (15-20% experience crisis)
- Dyspnoea: Exertional initially; orthopnoea (diaphragm weakness)
- Weak cough: Unable to clear secretions
- Respiratory failure: Myasthenic crisis—requires ventilatory support
Key Feature: Fluctuation and Fatigability
The hallmark of MG is fluctuating weakness with characteristic patterns:
- Worsens with activity: Repetitive muscle use causes progressive weakness
- Improves with rest: Brief rest (minutes) partially restores strength
- Diurnal variation: Worse at end of day; better after sleep
- Variability: Day-to-day and within-day fluctuation
- No pain: Weakness is painless (distinguishes from inflammatory myopathy)
- No sensory symptoms: Pure motor disorder
Clinical Classification (MGFA)
The Myasthenia Gravis Foundation of America (MGFA) Clinical Classification is the standard prognostic and therapeutic stratification system. [16]
| Class | Description | Features |
|---|---|---|
| I | Ocular only | Ptosis and/or diplopia; no other weakness |
| II | Mild generalised | IIa: Limb/axial predominant; IIb: Bulbar/respiratory predominant |
| III | Moderate generalised | IIIa: Limb/axial predominant; IIIb: Bulbar/respiratory predominant |
| IV | Severe generalised | IVa: Limb/axial predominant; IVb: Bulbar/respiratory predominant (feeding tube or severe dysphagia) |
| V | Intubation required | With or without mechanical ventilation (crisis) |
Post-Intervention Status:
- Pharmacologic remission: No symptoms on medication for ≥1 year
- Complete stable remission: No symptoms, no medication for ≥1 year
- Minimal manifestations: Subjective weakness but no functional limitations
- Improved, unchanged, worse: Compared to baseline
Red Flags: Myasthenic Crisis
Myasthenic crisis is defined as respiratory failure requiring intubation or non-invasive ventilation (NIV). Occurs in 15-20% of MG patients, usually within first 2-3 years of diagnosis. Mortality less than 5% with modern ICU care but requires early recognition and urgent intervention. [5]
Predictors of Crisis
- Oropharyngeal weakness (strongest predictor)
- MuSK-positive serology
- Thymoma
- Recent diagnosis (less than 2 years)
- Previous crisis (20-30% recurrence rate)
- Respiratory infection (most common trigger—30%)
- Recent surgery or medication changes
Clinical Features
- Respiratory: Dyspnoea, tachypnoea, use of accessory muscles, inability to cough, orthopnoea
- Bulbar: Severe dysphagia, pooling secretions, aspiration risk
- Objective measures:
- FVC less than 20mL/kg or less than 1L (normal ~4-5L)
- Negative inspiratory force (NIF) < -30cmH₂O (normal < -60cmH₂O)
- Unable to count to 20 in single breath
- Oxygen desaturation (late sign—do not wait for hypoxia)
Triggers (identifiable in 60-70%)
- Infection (30%): Respiratory most common
- Medications: Aminoglycosides, fluoroquinolones, beta-blockers
- Surgery
- Pregnancy/postpartum
- Tapering immunosuppression too rapidly
- Thyroid dysfunction
- No identifiable trigger in 30-40%
[!CAUTION] Myasthenic Crisis - Emergency Management
- ABCs: Airway protection, consider early intubation if FVC less than 15mL/kg, bulbar weakness, or declining trend
- ICU admission: All patients with impending or manifest crisis
- Stop pyridostigmine: Aids distinction from cholinergic crisis; restart after stabilisation
- Treat trigger: Antibiotics for infection (avoid contraindicated agents); discontinue offending drugs
- Acute immunotherapy: Plasmapheresis (5 exchanges over 10-14 days) OR IVIG (2g/kg over 2-5 days)
- Maintenance immunosuppression: Initiate or optimise corticosteroids and steroid-sparing agents
- Monitor: Serial FVC (every 4-6 hours), NIF, bulbar function, aspiration risk
5. Clinical Examination
Inspection
- Ptosis: Measure palpebral fissure; assess symmetry; observe for Cogan's lid twitch (overshoot on refixation)
- Facial appearance: Expressionless facies; "myasthenic snarl" (horizontal smile without elevation)
- Head posture: Head drop if severe neck flexor weakness
- Respiratory distress: Use of accessory muscles, paradoxical abdominal breathing
Fatigability Testing (Essential)
Ocular
- Sustained upgaze test: Patient looks upward for 60 seconds; observe for progressive ptosis (> 2mm decline = positive)
- Prolonged lateral gaze: Induces diplopia in 30-60 seconds if present
- Repeated eye closure: Fatigue of orbicularis oculi
Bulbar
- Counting test: Count aloud to 50; voice becomes nasal/dysarthric/weak
- Prolonged phonation: Sustain "eeee" for 30 seconds; voice fades
- Repetitive swallowing: Progressive difficulty
Limb
- Repeated shoulder abduction: 20 repetitions; progressive weakness
- Arm outstretched: Hold arms extended for 60 seconds; observe for drift
- Repeated sit-to-stand: 5 repetitions; progressive difficulty
Neck
- Sustained head lift: Supine patient lifts head off bed for 60 seconds; observe for fatigue/drop
Cranial Nerve Examination
- Eye movements: Complex ophthalmoplegia (no single nerve distribution); variable; spares pupils
- Facial nerve: Weak eye closure (cannot bury eyelashes); weak smile
- Bulbar: Weak palate elevation (nasal speech); weak tongue protrusion (rare)
Motor Examination
- Pattern: Proximal > distal; upper limbs commonly affected (deltoids, triceps)
- Tone: Normal (distinguishes from UMN and extrapyramidal disorders)
- Power: Medical Research Council (MRC) grading; document fatigability
- Reflexes: Normal or brisk (preserved—key differentiating feature from LMN disorders)
Sensory and Coordination
- Sensory: Normal (pure motor disorder)
- Coordination: Normal (unless weakness interferes)
Respiratory Assessment
- Forced Vital Capacity (FVC): Portable spirometry; normal ~4-5L; less than 20mL/kg or less than 1L indicates crisis
- Negative Inspiratory Force (NIF): < -30cmH₂O indicates respiratory muscle weakness
- Single breath count: Unable to count to 20 = significant weakness
6. Differential Diagnosis
MG can mimic various neurological and systemic conditions. Key to diagnosis is recognising fatigable weakness pattern, absence of sensory findings, and normal reflexes.
| Differential | Key Distinguishing Features |
|---|---|
| Lambert-Eaton Myasthenic Syndrome (LEMS) | Proximal weakness improving with exercise (facilitation); reduced/absent reflexes that augment post-exercise; autonomic features (dry mouth, constipation); associated with small-cell lung cancer (60%); antibodies to presynaptic voltage-gated calcium channels; incremental response on high-frequency (20-50Hz) RNS [17] |
| Botulism | Acute onset; descending paralysis (cranial → limbs); fixed dilated pupils (differentiates from MG); history of contaminated food/wound; toxin assay positive |
| Motor Neuron Disease (MND/ALS) | Progressive weakness without fluctuation or fatigue; fasciculations; mixed UMN and LMN signs; bulbar involvement (but no fluctuation); no improvement with rest; EMG shows fibrillation and fasciculation potentials |
| Chronic Progressive External Ophthalmoplegia (CPEO) | Slowly progressive bilateral ptosis and ophthalmoplegia; no diurnal variation; associated with mitochondrial disease; muscle biopsy shows ragged red fibres |
| Ocular myopathies | Non-fatigable weakness; no response to edrophonium or ice test; may have associated systemic features (e.g., thyroid eye disease) |
| Brainstem stroke | Sudden onset; sensory involvement; associated long tract signs; MRI brain shows acute infarct |
| Miller Fisher Syndrome | Ataxia, areflexia, ophthalmoplegia triad; preceding infection; anti-GQ1b antibodies; no fatigability |
| Polymyositis/Dermatomyositis | Proximal weakness without fatigability; muscle pain/tenderness; elevated CK (often > 1000); EMG shows myopathic changes; muscle biopsy diagnostic |
| Congenital Myasthenic Syndromes | Onset in infancy/childhood; genetic mutations in NMJ proteins; antibody-negative; positive family history; no autoimmune features |
| Thyroid eye disease | Proptosis; restrictive ophthalmoplegia (tethering); lid retraction (vs. ptosis); thyroid function abnormal |
Diagnostic Approach to Suspected MG
- Clinical suspicion: Fatigable weakness, ocular/bulbar symptoms, normal reflexes/sensation
- Bedside tests: Ice pack test, sustained upgaze, repetitive strength testing
- Antibody testing: AChR, MuSK, LRP4
- Electrophysiology: RNS (decremental response > 10%), SF-EMG (increased jitter)
- Imaging: CT/MRI thorax for thymoma
- Exclusion: Thyroid function, inflammatory markers, imaging as needed
7. Investigations
Antibody Testing (First-Line Diagnostic Test)
Serological testing for NMJ antibodies is the diagnostic cornerstone.
| Antibody | Sensitivity | Specificity | Clinical Associations |
|---|---|---|---|
| AChR (binding) | 85% (generalised); 50% (ocular) | > 95% | Classic MG; thymic hyperplasia or thymoma; good treatment response |
| MuSK | 5-8% (if AChR-negative) | > 99% | Female predominance; bulbar/neck/respiratory weakness; muscle atrophy; poor pyridostigmine response; excellent rituximab response [3,9] |
| LRP4 | 1-3% (double seronegative) | > 95% | Milder phenotype; similar to AChR-MG clinically |
| Striational (anti-titin, anti-ryanodine) | 30% (LOMG with thymoma) | Non-specific | Marker of thymoma, especially if age > 60; not diagnostic of MG alone |
Testing Strategy
- First-line: AChR antibodies (binding assay)
- If negative and clinical suspicion remains: MuSK antibodies
- If both negative: LRP4 antibodies
- In late-onset MG: Consider striational antibodies as thymoma marker
Seronegative MG (10%)
- No detectable antibodies by standard assays
- May have low-affinity AChR antibodies detectable by cell-based assays
- Diagnosis based on clinical features + electrophysiology + treatment response
- Clinical course similar to AChR-positive disease
Electrophysiology
Repetitive Nerve Stimulation (RNS)
- Technique: Supramaximal stimulation of motor nerve (e.g., ulnar, facial) at 2-5Hz; record compound muscle action potential (CMAP)
- Positive finding: > 10% decrement in CMAP amplitude between 1st and 4th/5th stimulus
- Sensitivity: 50-60% (generalised MG); 10-30% (ocular MG)—improves to 80% if proximal muscles tested (trapezius, facial)
- Specificity: High (> 95%) if > 10% decrement observed
- Mechanism: Depleted ACh stores unmask reduced safety factor
Single-Fibre EMG (SF-EMG)
- Technique: Measures neuromuscular jitter (variability in interpotential interval between two muscle fibres in same motor unit)
- Positive finding: Increased jitter (>mean normal for muscle) with or without blocking
- Sensitivity: 95-99% (most sensitive test for MG)
- Specificity: Moderate—increased jitter occurs in other NMJ disorders (LEMS, botulism) and myopathies
- Utility: Confirms NMJ disorder when antibodies negative; not specific for MG
Ice Pack Test (Bedside)
- Indication: Ocular MG with ptosis
- Technique: Apply ice pack or ice-filled glove to ptotic eyelid for 2 minutes; measure palpebral fissure before and after
- Positive test: ≥2mm improvement in ptosis
- Sensitivity: 80-90% in ocular MG with ptosis [12]
- Mechanism: Cooling slows AChE activity, increasing ACh availability at NMJ
- Advantage: Non-invasive, immediate, no cost
Edrophonium (Tensilon) Test (Rarely Used Now)
Historically the bedside diagnostic test, now largely replaced by safer alternatives (ice test, antibody testing).
- Technique: IV edrophonium (short-acting AChE inhibitor) 2mg test dose, then 8mg if no reaction
- Positive: Transient improvement in weakness (ptosis, diplopia) within 30-60 seconds
- Risks: Bradycardia, bronchospasm, syncope (requires atropine available and cardiac monitoring)
- Current role: Minimal—superseded by safer tests
Imaging
CT or MRI Thorax (Mandatory in All MG Patients)
- Indication: Detect thymoma (10-20% of AChR-positive MG) [6]
- Timing: At diagnosis
- Findings:
- "Thymic hyperplasia: Enlarged thymus with normal architecture; germinal centres on pathology"
- "Thymoma: Anterior mediastinal mass; heterogeneous enhancement; potential invasion (WHO classification: A, AB, B1-B3, C)"
- "Normal/atrophic: Common in late-onset MG (> 60 years)"
- Follow-up: Repeat imaging not routinely needed unless thymoma or clinical change
WHO Classification of Thymoma (Relevant for Oncological Management)
- Type A: Medullary, spindle cells—best prognosis
- Type AB: Mixed
- Type B1-B3: Cortical, increasing atypia—worse prognosis
- Type C: Thymic carcinoma—aggressive
Pulmonary Function Tests
- Forced Vital Capacity (FVC): Baseline and monitoring; less than 20mL/kg indicates crisis risk
- Negative Inspiratory Force (NIF): < -30cmH₂O suggests respiratory muscle weakness
- Indications: Baseline in all generalised MG; serial monitoring if bulbar or respiratory symptoms; urgent if suspected crisis
Additional Investigations
- Thyroid function tests (TFTs): 5-10% of MG patients have autoimmune thyroid disease (Graves' or Hashimoto's)
- Inflammatory markers: Usually normal; exclude inflammatory myopathy if atypical features
- Creatine kinase (CK): Normal in MG (elevated in myositis)
- CT chest for malignancy: If LEMS suspected (associated with small-cell lung cancer)
8. Management
Management of MG is multimodal, combining symptomatic treatment (acetylcholinesterase inhibitors), immunosuppression (corticosteroids and steroid-sparing agents), thymectomy in selected patients, and acute therapies (plasmapheresis, IVIG) for exacerbations and crisis. Treatment is tailored to antibody subtype, disease severity, and patient factors. [1,13]
Symptomatic Treatment: Acetylcholinesterase Inhibitors
Pyridostigmine
- Mechanism: Inhibits acetylcholinesterase in synaptic cleft → increased ACh availability → improved neuromuscular transmission
- Indications: First-line for all MG patients; provides symptomatic relief without disease modification
- Dosing: Start 30-60mg three to four times daily; titrate to effect; maximum 120mg four times daily (total 480mg/day)
- Onset: 30-60 minutes; duration 3-4 hours
- Efficacy: Effective in AChR-positive MG; poor response in MuSK-positive MG [9]
- Side effects (cholinergic):
- "GI: Diarrhoea, abdominal cramps, nausea (most common—30-40%)"
- "Muscarinic: Hypersalivation, lacrimation, miosis, bradycardia"
- "Nicotinic: Fasciculations, muscle cramps"
- Management of side effects: Reduce dose; take with food; consider antimuscarinic (loperamide for diarrhoea; avoid systemic antimuscarinics which cross blood-brain barrier)
- Limitations: Provides only partial symptom control in most patients; does not prevent disease progression
Cholinergic Crisis
Over-treatment with pyridostigmine (rare with standard dosing) causes excessive cholinergic stimulation:
- Features: Weakness, excessive secretions (salivation, lacrimation), miosis, bradycardia, diarrhoea, abdominal cramps
- Distinguish from myasthenic crisis: Parasympathetic features present; temporarily stop pyridostigmine—improvement confirms cholinergic cause
- Management: Stop pyridostigmine; supportive care; consider atropine if severe bradycardia
Immunosuppression
Most MG patients require long-term immunosuppression to achieve disease control and enable steroid tapering.
Corticosteroids (First-Line Immunosuppression)
Prednisolone/Prednisone
- Indications: Generalised MG inadequately controlled on pyridostigmine; ocular MG causing functional impairment
- Dosing:
- "Gradual initiation (preferred): Start 10mg daily; increase by 5mg every 3-5 days to 0.5-1mg/kg (max 60-80mg)"
- "High-dose initiation (if urgent control needed): 1mg/kg daily, but monitor closely for initial worsening"
- Onset: Improvement within 2-4 weeks; maximum benefit at 6-12 weeks
- Maintenance: Taper slowly once improvement achieved (reduce by 5mg per month); aim for minimum effective dose (often 5-15mg alternate days)
- Response rate: 70-80% achieve significant improvement
Critical Warning: Steroid-Induced Worsening
- Occurs in 30-50% if high-dose initiated abruptly (e.g., 60mg prednisolone)
- Typically 7-10 days after initiation
- Mechanism unclear (possibly immune activation or electrolyte shifts)
- Can precipitate myasthenic crisis requiring ICU admission
- Prevention: Start low (10mg), increase gradually; consider inpatient initiation if severe disease or respiratory involvement [1,13]
Side Effects (long-term steroids)
- Metabolic: Weight gain, diabetes, dyslipidaemia
- Bone: Osteoporosis (prophylaxis with calcium, vitamin D, bisphosphonates if high risk)
- GI: Peptic ulcer (consider PPI if risk factors)
- Ophthalmological: Cataracts, glaucoma
- Psychiatric: Mood changes, insomnia
- Infection risk
- Monitoring: Blood pressure, glucose, bone density, ophthalmology
Steroid-Sparing Agents (Second-Line)
Goal: Enable steroid dose reduction and minimise long-term steroid toxicity.
| Agent | Dose | Onset | Monitoring | Notes |
|---|---|---|---|---|
| Azathioprine | 2-3mg/kg/day (target 150-200mg) | 6-12 months | FBC, LFTs monthly initially, then 3-monthly; TPMT activity before starting (deficiency causes severe myelosuppression) | Most evidence-based steroid-sparing agent; malignancy risk increased (especially lymphoma); teratogenic [1,13] |
| Mycophenolate mofetil | 1-1.5g twice daily | 3-6 months | FBC, LFTs 3-monthly | Alternative to azathioprine; better GI tolerability; limited evidence in MG; teratogenic |
| Methotrexate | 15-25mg weekly (oral or subcutaneous) | 3-6 months | FBC, LFTs, renal function 3-monthly; CXR baseline | Limited evidence; used if azathioprine/mycophenolate contraindicated; folic acid supplementation 5mg weekly; teratogenic |
| Ciclosporin | 3-5mg/kg/day in divided doses | 2-4 months | Renal function, BP, levels 3-monthly | Effective but significant toxicity (nephrotoxicity, hypertension); reserved for refractory cases |
| Tacrolimus | 3-5mg twice daily | 2-4 months | Renal function, glucose, levels 3-monthly | Similar to ciclosporin; possibly better tolerability |
Rituximab (B-Cell Depleting Therapy)
- Mechanism: Monoclonal anti-CD20 antibody → B-cell depletion → reduced antibody production
- Indications:
- "MuSK-positive MG: Excellent response (80-90% significant improvement); emerging as first-line after steroids [9]"
- "AChR-positive MG: Moderate response (50-60% improvement); reserved for refractory cases (failed ≥2 immunosuppressants) or steroid-dependent disease"
- "Seronegative MG: Variable response"
- Dosing:
- "Lymphoma protocol: 375mg/m² weekly × 4 weeks"
- "Rheumatoid arthritis protocol: 1000mg × 2 doses, 2 weeks apart (increasingly used—lower cost, similar efficacy)"
- Onset: 1-6 months; duration of effect 6-24 months (retreatment often needed)
- Monitoring: B-cell depletion (CD19/CD20 count); immunoglobulin levels; infection surveillance
- Side effects: Infusion reactions (premedicate with antihistamine, paracetamol, steroid); infection risk (especially respiratory); progressive multifocal leukoencephalopathy (rare); hypogammaglobulinaemia
- Evidence: Multiple observational studies; ongoing RCTs (BeatMG trial) [9]
Newer Agents (Emerging Therapies)
- Eculizumab: Monoclonal anti-C5 antibody blocking complement; FDA-approved for refractory generalised AChR-positive MG (REGAIN trial showed benefit); very high cost; requires meningococcal vaccination
- Efgartigimod: Neonatal Fc receptor antagonist reducing IgG (including pathogenic antibodies); FDA-approved 2021; administered as IV infusion cycles
- Rozanolixizumab: Fc receptor antagonist; similar mechanism to efgartigimod
Thymectomy
Surgical removal of thymus is an established treatment for MG, with benefits including improved disease control and reduced immunosuppression requirements.
Indications [1,7]
- Thymoma: Mandatory (oncological indication; 30-50% of thymomas are locally invasive or malignant)
- Generalised AChR-positive MG, age 18-65: Recommended based on MGTX trial [7]
- Ocular MG: Consider if progressing or impaired quality of life
- MuSK-positive MG: No benefit—thymus not involved in pathogenesis [9]
MGTX Trial (2016) [7]
Landmark randomised controlled trial definitively proving thymectomy benefit in non-thymomatous MG.
- Design: 126 patients (age 18-65, generalised AChR-positive MG, disease less than 5 years) randomised to extended transsternal thymectomy + prednisone vs. prednisone alone
- Results (3-year follow-up):
- Lower disease severity (QMG score 6.15 vs. 8.99, pless than 0.001)
- Lower prednisone requirement (44mg vs. 60mg alternate-day average, pless than 0.001)
- Fewer patients requiring additional immunosuppression
- Conclusion: Thymectomy improves outcomes in non-thymomatous generalised AChR-positive MG aged 18-65
Surgical Approaches
- Extended transsternal thymectomy: Gold standard (used in MGTX trial); complete thymic tissue removal including anterior mediastinal fat
- VATS (video-assisted thoracoscopic surgery): Minimally invasive; shorter recovery; comparable efficacy in experienced hands
- Robotic thymectomy: Emerging; comparable outcomes to VATS
Timing
- Thymoma: Urgent (oncological priority)
- Non-thymomatous: Elective; optimise medical control before surgery to reduce perioperative risk
Outcomes
- Improvement: 60-70% at 5 years
- Remission: 20-40% (medication-free) at long-term follow-up
- Benefit may take 2-5 years to manifest fully
- No benefit in MuSK-positive MG
Acute Immunotherapy (Myasthenic Crisis and Severe Exacerbations)
Plasmapheresis (Plasma Exchange, PLEX)
- Mechanism: Removes circulating antibodies (IgG) via extracorporeal plasma separation
- Indications:
- Myasthenic crisis
- Severe exacerbation
- Pre-operative optimisation (e.g., before thymectomy in severe disease)
- Protocol: 5 exchanges over 10-14 days; each exchange removes 1-1.5 plasma volumes (~3-4L)
- Onset: Improvement within 1-3 days
- Duration: 4-8 weeks (antibody levels rebound)
- Efficacy: 75-85% respond
- Complications: Line-related infection/thrombosis, hypotension, citrate toxicity (hypocalcaemia), coagulopathy (factor depletion)
- Advantages: Rapid onset; no immunosuppression (safe in infection)
Intravenous Immunoglobulin (IVIG)
- Mechanism: Multiple (Fc receptor blockade, complement inhibition, anti-idiotypic antibodies)
- Indications: Same as plasmapheresis
- Dosing: 2g/kg total dose over 2-5 days (e.g., 0.4g/kg/day × 5 days)
- Onset: Improvement within 3-7 days (slower than PLEX)
- Duration: 4-8 weeks
- Efficacy: 70-80% respond (non-inferior to PLEX in most studies) [10]
- Complications: Headache, aseptic meningitis, thrombotic events (stroke, MI), renal impairment, volume overload, haemolysis
- Advantages: Easier administration than PLEX; no vascular access required; fewer complications
PLEX vs. IVIG: Comparable efficacy; choice based on availability, patient factors (PLEX contraindicated if poor vascular access, coagulopathy, or haemodynamic instability), and institutional preference. [10]
Management of Myasthenic Crisis
Definition: Respiratory failure requiring mechanical ventilation (invasive or non-invasive).
ICU Management
-
Airway and Ventilation:
- Intubate if FVC less than 15mL/kg, NIF < -30cmH₂O, hypoxia, or declining trend
- NIV may be considered in selected patients (alert, cooperative, no severe bulbar dysfunction)
- Monitor FVC every 4-6 hours; intubate before precipitous decline
-
Stop Pyridostigmine:
- Temporarily discontinue to exclude cholinergic contribution
- Restart at lower dose once stabilised
-
Identify and Treat Trigger:
- Infection (most common): Antibiotics (avoid aminoglycosides, fluoroquinolones if possible)
- Medications: Discontinue offending agents
- Surgery, pregnancy, thyroid dysfunction
-
Acute Immunotherapy:
- Plasmapheresis (preferred if available): 5 exchanges over 10-14 days
- OR IVIG: 2g/kg over 2-5 days
- Comparable efficacy; PLEX faster onset [10]
-
Initiate/Optimise Long-Term Immunosuppression:
- Start or increase prednisolone (low-dose initiation if not already on steroids)
- Consider steroid-sparing agent (azathioprine, mycophenolate)
-
Supportive Care:
- DVT prophylaxis, stress ulcer prophylaxis
- Nutrition (NG/PEG if prolonged)
- Physiotherapy
-
Weaning:
- Most patients wean from ventilation within 2-4 weeks
- Predictors of successful weaning: FVC > 20mL/kg, NIF < -40cmH₂O, resolution of trigger
Prognosis: Mortality less than 5% with modern ICU care; recurrence risk 20-30%. [5]
Special Populations
Pregnancy
- MG unpredictable in pregnancy: 30% improve, 30% worsen (especially postpartum), 40% unchanged
- Management:
- Continue pyridostigmine (safe in pregnancy)
- Prednisolone safe (first-line immunosuppression)
- "Azathioprine: Generally continued if already on (stopping risks exacerbation); teratogenic risk low in practice"
- Stop methotrexate, mycophenolate (teratogenic)
- IVIG safe for exacerbations
- Delivery: Vaginal preferred; avoid magnesium sulphate (worsens MG); monitor for crisis postpartum
- Neonatal MG: 10-20% of infants born to AChR-positive mothers develop transient MG (antibody transfer); self-limiting (resolves by 3 months)
Elderly (> 65 years)
- Late-onset MG increasing incidence
- More severe disease; lower remission rates
- Thymectomy benefit unclear (excluded from MGTX trial); consider if less than 70 and good functional status
- Steroid side effects more problematic (osteoporosis, diabetes, infection)
- Comorbidities complicate management
Drugs to AVOID in Myasthenia Gravis
Multiple medications can worsen neuromuscular transmission and precipitate exacerbations or crisis. [14]
| Category | Agents | Mechanism |
|---|---|---|
| Antibiotics | Aminoglycosides (gentamicin, tobramycin), fluoroquinolones (ciprofloxacin, levofloxacin), macrolides (azithromycin, erythromycin), tetracyclines | Presynaptic ACh release inhibition; postsynaptic receptor blockade |
| Cardiac | Beta-blockers (propranolol, atenolol), calcium channel blockers (verapamil, diltiazem), quinidine, procainamide | Impaired neuromuscular transmission |
| Neuromuscular blockers | Succinylcholine, rocuronium, vecuronium | Prolonged paralysis (use with extreme caution; reduce dose) |
| Magnesium | IV magnesium sulphate, magnesium-containing laxatives | Presynaptic ACh release inhibition |
| Other | D-penicillamine (induces MG), botulinum toxin, quinine, chloroquine, statins (rarely) | Various mechanisms |
Clinical Pearl: Always screen medications before prescribing in MG patients. Use alternative antibiotics (e.g., penicillins, cephalosporins) when possible. If no alternative exists, use with caution and close monitoring.
9. Complications
| Complication | Incidence | Risk Factors | Prevention | Management |
|---|---|---|---|---|
| Myasthenic crisis | 15-20% lifetime | Bulbar weakness, MuSK-positive, thymoma, infection, recent diagnosis | Optimise immunosuppression; avoid triggers; monitor FVC if respiratory symptoms | ICU, PLEX/IVIG, mechanical ventilation, treat trigger [5] |
| Aspiration pneumonia | Common if bulbar weakness | Dysphagia, weak cough | Swallow assessment; modified diet; upright positioning; NBM if unsafe | Antibiotics; aspiration precautions; consider NG/PEG |
| Respiratory failure | 15-20% (crisis) | As above | As above | Intubation; ventilation; PLEX/IVIG |
| Thymoma-related | 10-20% have thymoma; 30-50% of thymomas invade locally | Late-onset MG; high anti-titin antibodies | CT thorax at diagnosis | Surgical resection; oncological staging; adjuvant radiotherapy/chemotherapy if invasive |
| Steroid complications | Variable (dose and duration dependent) | Long-term high-dose corticosteroids | Steroid-sparing agents; osteoporosis prophylaxis (calcium, vitamin D, bisphosphonates); PPI if GI risk; glucose/BP monitoring | Treat specific complications; taper steroids |
| Immunosuppression-related infections | Increased risk | Combination immunosuppression; high-dose steroids | Vaccinations (pneumococcal, influenza, shingles—live vaccines contraindicated if on immunosuppression); prophylaxis (PJP if high risk) | Prompt treatment; consider immunosuppression reduction |
| Treatment-refractory disease | 10-30% inadequately controlled | MuSK-positive; thymoma; late-onset | Optimise combination therapy; consider rituximab or newer agents | Rituximab, eculizumab, efgartigimod; chronic IVIG |
| Psychosocial impact | Common | Chronic disease; fluctuating symptoms; functional limitation | Patient education; support groups; psychological support | Counselling; occupational therapy; disability assessment if needed |
10. Prognosis
Historical Context
Pre-1950s (before acetylcholinesterase inhibitors and immunosuppression): Mortality 30-40%, primarily from respiratory failure and aspiration.
Modern era (with ICU care, immunosuppression, PLEX/IVIG): Mortality less than 5%, near-normal life expectancy for most patients. [5]
Outcomes with Treatment
Disease Control
- Remission (medication-free, asymptomatic ≥1 year): 20-30% achieve complete stable remission [4]
- Pharmacological remission (asymptomatic on medication): 30-50%
- Minimal manifestations (subjective weakness without functional limitation): 20-30%
- Refractory disease (inadequate control despite ≥2 immunosuppressants): 10-30%
Mortality
- Overall: less than 5% in modern cohorts [5]
- Crisis-related: less than 5% mortality during crisis (down from 40% pre-ICU era)
- Driven by: Age (elderly worse), comorbidities (cardiac, respiratory), thymoma (invasive disease)
Quality of Life
- Significant impairment in untreated or poorly controlled disease
- Improvement with treatment correlates with disease control
- Persistent fatigue common even in remission (40-60%)
- Steroid side effects impact QoL (weight gain, mood, osteoporosis)
Prognostic Factors
Favourable Prognosis
- Early-onset MG (age less than 40, especially women)
- AChR-positive
- Thymic hyperplasia (vs. thymoma or normal)
- Ocular-only disease (though 50-80% generalise within 2 years)
- Early treatment initiation
- Thymectomy in appropriate patients
- Good treatment response
Poor Prognosis
- Late-onset MG (age > 60)
- MuSK-positive (more severe bulbar/respiratory disease, though rituximab response excellent) [9]
- Thymoma (especially invasive)
- Bulbar or respiratory weakness at presentation
- Delayed diagnosis/treatment
- Refractory to conventional immunosuppression
- Multiple comorbidities
Natural History by Subtype
Ocular MG
- 15-20% remain purely ocular
- 50-80% generalise within 2 years (majority in first year)
- If purely ocular at 2 years, low risk of subsequent generalisation (less than 10%)
Generalised AChR-Positive MG
- With treatment: 70-80% achieve good disease control
- Crisis risk highest in first 2-3 years
- Long-term outcomes good with modern therapy
MuSK-Positive MG
- More severe disease; higher crisis rate
- Excellent response to rituximab (80-90%) [9]
- Poorer response to conventional immunosuppression and pyridostigmine
- No thymectomy benefit
Thymomatous MG
- MG often more severe
- Thymoma prognosis depends on WHO classification and staging (Masaoka)
- Post-thymectomy: MG may improve, remain stable, or (rarely) worsen
Long-Term Follow-Up
- Lifelong monitoring required (even in remission—relapse possible)
- Clinic review: Every 3-12 months depending on disease control
- Monitoring: Disease severity (QMG, ADL scores), medication side effects, FVC if respiratory involvement
- Imaging: Repeat CT thorax not routinely needed unless thymoma or clinical change
- Treatment adjustments: Taper immunosuppression cautiously in stable patients; aim for minimal effective dose
11. Prevention and Screening
Primary Prevention
No established primary prevention strategies (aetiology involves genetic susceptibility and unclear environmental triggers).
Modifiable Risk Factors
- Smoking: Emerging evidence suggests association with increased MG risk (Brinkman Index correlation with severity) [18]
- Infection control: Respiratory infections are common crisis triggers—influenza and pneumococcal vaccination recommended
Secondary Prevention (Preventing Exacerbations and Crisis)
Patient Education
- Recognise warning signs of exacerbation (increasing ptosis, diplopia, dysphagia, dyspnoea)
- Avoid trigger medications (provide written list)
- Seek prompt treatment for infections
- Medication compliance
Medical Optimisation
- Adequate immunosuppression to achieve disease control
- Regular monitoring (clinical, FVC if respiratory involvement)
- Treat comorbidities (thyroid disease, autoimmune conditions)
Crisis Prevention
- Optimise control before elective surgery
- Plasmapheresis or IVIG pre-operatively if high risk
- Avoid respiratory depressants and neuromuscular blockers (or use cautiously with reduced dosing)
- Postpartum monitoring (high-risk period for exacerbation)
Screening
Thymoma Screening
- All MG patients: CT/MRI thorax at diagnosis [6]
- No routine surveillance imaging if initial scan normal (unless clinical change)
Neonatal Screening
- Infants born to AChR-positive mothers: Monitor for transient neonatal MG (10-20% incidence)
- Features: Hypotonia, weak cry, feeding difficulty, respiratory distress
- Self-limiting (resolves by 3 months as maternal antibodies clear)
12. Key Guidelines and Evidence
International Guidelines
International Consensus Guidance for Management of Myasthenia Gravis (2016) [1]
Task Force convened by Myasthenia Gravis Foundation of America; 15 international experts; RAND/UCLA appropriateness methodology.
Key Recommendations:
- Symptomatic treatment: Pyridostigmine first-line
- Immunosuppression: Corticosteroids first-line; azathioprine or mycophenolate as steroid-sparing agents
- Thymectomy: Recommended for generalised AChR-positive MG age 18-65 (supported by MGTX trial)
- Acute therapy: PLEX or IVIG for crisis and severe exacerbations (equivalent efficacy)
- MuSK-MG: Rituximab emerging as preferred second-line; no thymectomy benefit
- Pregnancy: Pyridostigmine and prednisolone safe; IVIG for exacerbations
AANEM (American Association of Neuromuscular & Electrodiagnostic Medicine) Guidelines
Diagnosis of MG: Antibody testing (AChR, MuSK, LRP4), electrophysiology (RNS, SF-EMG), clinical criteria.
Landmark Trials
MGTX Trial (2016) [7]
Randomized Trial of Thymectomy in Myasthenia Gravis
- Design: RCT, 126 patients (age 18-65, generalised AChR-positive MG, disease less than 5 years, MGFA class II-IV), extended transsternal thymectomy + prednisone vs. prednisone alone
- Primary outcomes: Time-weighted average QMG score and prednisone dose over 3 years
- Results:
- "Thymectomy group: Lower QMG (6.15 vs. 8.99, pless than 0.001), lower prednisone dose (44mg vs. 60mg, pless than 0.001)"
- Fewer patients required additional immunosuppression
- Conclusion: Thymectomy improves outcomes in non-thymomatous generalised AChR-positive MG age 18-65
- Citation: Wolfe GI, et al. N Engl J Med. 2016;375(6):511-522. PMID: 27509100
REGAIN Trial (2017)
Eculizumab (anti-C5 complement inhibitor) in refractory generalised AChR-positive MG: showed benefit, leading to FDA approval for refractory MG.
BeatMG Trial (Ongoing)
RCT of rituximab in MG; results pending.
Key Reviews and Meta-Analyses
Gilhus NE, et al. Lancet Neurol (2015) [3]
Myasthenia gravis: subgroup classification and therapeutic strategies
Comprehensive review of MG subgroups (AChR, MuSK, LRP4, seronegative, ocular, thymoma-associated, early/late-onset) and tailored treatment strategies.
Sanders DB, et al. Neurology (2016) [1]
International consensus guidance for management of myasthenia gravis
Formal consensus using RAND/UCLA methodology; comprehensive treatment guidance.
Gilhus NE, et al. Nat Rev Dis Primers (2019) [19]
Myasthenia gravis
Comprehensive disease primer covering epidemiology, pathophysiology, diagnosis, treatment, and prognosis.
Claytor B, Lowry TJ. Muscle Nerve (2023) [5]
Myasthenic crisis
Comprehensive review: definition, epidemiology (15-20% incidence), triggers, risk factors, management (PLEX vs. IVIG), outcomes (mortality less than 5%).
Evidence Gaps and Ongoing Research
- Optimal sequencing of immunosuppressants
- Role of rituximab in AChR-positive MG (awaiting BeatMG trial results)
- New agents: efgartigimod, rozanolixizumab (recently approved; long-term data needed)
- Thymectomy in late-onset MG (> 65 years)—MGTX excluded this group
- Biomarkers predicting treatment response
- Precision medicine approaches based on antibody subtype and genotype
13. Viva and Examination Focus
Opening Statement for Viva
"Myasthenia gravis is a chronic autoimmune disorder of the neuromuscular junction characterised by fluctuating fatigable weakness that worsens with activity and improves with rest. It is mediated by antibodies against postsynaptic proteins, most commonly the acetylcholine receptor in 80-85% of cases, muscle-specific kinase in 5-8%, and LRP4 in less than 1%. Clinically, patients present with ptosis, diplopia, bulbar weakness, and proximal limb weakness. Diagnosis is confirmed by antibody testing, electrophysiology showing decremental response on repetitive nerve stimulation, and CT thorax to detect thymoma in 10-20% of cases. Treatment involves symptomatic therapy with pyridostigmine, immunosuppression with corticosteroids and steroid-sparing agents such as azathioprine, and thymectomy for thymomatous or generalised AChR-positive disease under age 65, as demonstrated by the MGTX trial. Myasthenic crisis, occurring in 15-20% of patients, requires ICU management with plasmapheresis or IVIG and mechanical ventilation. With modern treatment, mortality is less than 5% and prognosis is excellent."
High-Yield Facts for Examinations
Epidemiology
- Incidence 8-30 per million; prevalence 150-250 per million [11]
- Bimodal age distribution: women 20-40, men 60-80 [4,11]
- 15-20% experience myasthenic crisis [5]
Pathophysiology
- AChR antibodies (80-85%): IgG1/IgG3, complement-mediated destruction, receptor internalisation [2]
- MuSK antibodies (5-8%): IgG4, non-complement fixing, disrupts AChR clustering [3]
- Thymic hyperplasia (60-70%), thymoma (10-20%) [6]
Clinical Features
- Fatigable weakness worsening with activity, improving with rest (pathognomonic)
- Ocular (85% at presentation): Ptosis, diplopia
- Bulbar (60%): Dysarthria, dysphagia
- Normal reflexes and sensation (key differentiating feature)
Diagnosis
- AChR antibodies: 85% sensitivity (generalised), 50% (ocular)
- RNS: > 10% decrement at 2-5Hz (50-60% sensitivity)
- SF-EMG: Increased jitter (95-99% sensitivity, most sensitive test)
- Ice pack test: ≥2mm ptosis improvement (80-90% sensitivity in ocular MG) [12]
- CT/MRI thorax: Mandatory to detect thymoma [6]
Treatment
- Pyridostigmine: 30-120mg QDS (poor response in MuSK-MG)
- Prednisolone: Start LOW (10mg), increase gradually to avoid worsening [1,13]
- Azathioprine: 2-3mg/kg, onset 6-12 months, check TPMT first
- Thymectomy: Mandatory for thymoma; recommended for generalised AChR-positive MG age 18-65 (MGTX trial) [7]
- MuSK-MG: Rituximab highly effective (80-90% response) [9]
- Crisis: PLEX (5 exchanges) or IVIG (2g/kg); PLEX faster onset [10]
Crisis Predictors and Management
- Risk factors: Bulbar weakness, MuSK-positive, thymoma, respiratory infection
- ICU admission, intubate if FVC less than 15mL/kg
- Stop pyridostigmine temporarily
- PLEX or IVIG; treat trigger [5]
Drugs to Avoid
- Aminoglycosides, fluoroquinolones, beta-blockers, magnesium, D-penicillamine [14]
Prognosis
- Mortality less than 5% (modern era) [5]
- 20-30% achieve medication-free remission [4]
Common Exam Questions and Model Answers
Q1: How would you investigate a patient with suspected myasthenia gravis?
Model Answer: "I would approach this systematically with clinical assessment, serology, electrophysiology, and imaging.
First, clinical assessment: Detailed history focusing on fatigable weakness, diurnal variation, and examination demonstrating fluctuating weakness with sustained upgaze and repetitive shoulder abduction tests.
Second, bedside tests: Ice pack test for ptosis—applying ice for 2 minutes should improve ptosis by at least 2mm if MG is present, with 80-90% sensitivity.
Third, serology: AChR antibodies first-line, present in 85% of generalised MG and 50% of ocular MG. If negative, I would test for MuSK antibodies (5-8% of cases) and LRP4 antibodies (1-3%).
Fourth, electrophysiology: Repetitive nerve stimulation at 2-5Hz typically shows greater than 10% decremental response with 50-60% sensitivity. Single-fibre EMG showing increased jitter is the most sensitive test at 95-99% but less specific.
Fifth, imaging: CT or MRI thorax is mandatory in all MG patients to detect thymoma, present in 10-20% of AChR-positive cases.
Finally, pulmonary function tests: Baseline FVC to assess respiratory muscle strength and monitor for crisis risk."
Q2: Describe your management approach to myasthenic crisis.
Model Answer: "Myasthenic crisis is defined as respiratory failure requiring mechanical ventilation, occurring in 15-20% of MG patients with mortality under 5% with modern ICU care.
My immediate priorities are airway management and stabilisation. I would assess respiratory function with FVC and negative inspiratory force, intubating if FVC is less than 15mL/kg, NIF less than -30cmH₂O, or if there is declining trend or bulbar weakness with aspiration risk.
I would admit the patient to ICU and temporarily stop pyridostigmine to exclude cholinergic contribution and aid diagnosis.
Next, I would identify and treat triggers—respiratory infection is most common in 30% of cases, requiring appropriate antibiotics while avoiding aminoglycosides and fluoroquinolones which can worsen MG. Other triggers include medications, surgery, and pregnancy.
For acute immunotherapy, I would use plasmapheresis—5 exchanges over 10-14 days—which is preferred as it has faster onset than IVIG. Alternatively, IVIG 2g/kg over 2-5 days is equally effective. Multiple studies show comparable efficacy between the two.
Simultaneously, I would initiate or optimise long-term immunosuppression with prednisolone, starting at low dose if not already on steroids to avoid steroid-induced worsening, and considering steroid-sparing agents like azathioprine or mycophenolate.
Most patients wean from ventilation within 2-4 weeks. I would monitor FVC serially and look for resolution of the precipitating trigger before attempting weaning."
Q3: What is the evidence for thymectomy in myasthenia gravis?
Model Answer: "The landmark MGTX trial published in the New England Journal of Medicine in 2016 definitively proved thymectomy benefit in non-thymomatous myasthenia gravis.
This was a multicentre randomised controlled trial of 126 patients aged 18-65 with generalised AChR-positive MG and disease duration less than 5 years, comparing extended transsternal thymectomy plus alternate-day prednisone versus prednisone alone.
The primary outcomes were time-weighted average Quantitative Myasthenia Gravis score and prednisone dose over 3 years. Results showed that patients who underwent thymectomy had significantly lower disease severity—QMG score 6.15 versus 8.99, p less than 0.001—and lower average prednisone requirement—44mg versus 60mg alternate-day, p less than 0.001. Fewer patients in the thymectomy group required additional immunosuppression.
Based on this evidence, international consensus guidelines now recommend thymectomy for generalised AChR-positive MG in patients aged 18-65. Thymectomy is mandatory for thymoma due to oncological considerations, as 30-50% of thymomas are locally invasive.
Importantly, thymectomy shows no benefit in MuSK-positive MG as thymic pathology is not part of the disease mechanism in this subgroup."
Q4: How do you distinguish between different causes of fatigable weakness?
Model Answer: "The key differentials for fatigable weakness are myasthenia gravis, Lambert-Eaton myasthenic syndrome, and congenital myasthenic syndromes, each with distinct features.
Myasthenia gravis presents with weakness worsening with activity and improving with rest, affecting ocular, bulbar, and proximal limb muscles. Reflexes and sensation are normal. AChR or MuSK antibodies are present in 90% of cases. Repetitive nerve stimulation shows decremental response at low frequency.
Lambert-Eaton myasthenic syndrome presents with proximal weakness that paradoxically improves with initial activity before fatiguing, due to facilitation. Reflexes are reduced or absent but augment after exercise. Autonomic features like dry mouth and constipation are common. It is associated with small-cell lung cancer in 60% of cases and antibodies to voltage-gated calcium channels. RNS shows incremental response at high frequency 20-50Hz.
Congenital myasthenic syndromes present in infancy or childhood with fluctuating weakness, have positive family history, are antibody-negative, and result from genetic mutations affecting neuromuscular junction proteins.
Botulism causes acute descending paralysis with fixed dilated pupils, which distinguishes it from MG, and has history of contaminated food or wound.
The clinical pattern, antibody testing, electrophysiology, and associated features allow differentiation."
Q5: What are the specific considerations in managing MuSK-positive myasthenia gravis?
Model Answer: "MuSK-positive MG comprises 5-8% of MG cases and has several distinctive features requiring tailored management.
Clinically, MuSK-positive patients show strong female predominance with ratio 9:1, predominantly bulbar and neck/respiratory weakness, facial and tongue atrophy, and minimal ocular involvement compared to AChR-positive disease. Crisis rates are higher.
Pathophysiologically, MuSK antibodies are IgG4 subclass, which are non-complement fixing, explaining the different phenotype. MuSK is essential for AChR clustering via the agrin-LRP4-MuSK pathway, and antibodies disrupt this function.
Regarding treatment response, MuSK-positive patients show poor response to pyridostigmine and acetylcholinesterase inhibitors, likely due to the different pathophysiology. Thymectomy provides no benefit as thymic pathology is not involved.
However, rituximab is highly effective in MuSK-positive MG, with 80-90% of patients showing significant improvement. This excellent response to B-cell depletion has led to rituximab being considered first-line after steroids in this subgroup, rather than reserving it for refractory cases as in AChR-positive disease.
Standard immunosuppression with prednisolone is still used but these patients often require higher doses and additional agents. The prognosis with appropriate treatment including rituximab is now much improved."
Common Mistakes That Fail Candidates
❌ Starting high-dose prednisolone (60mg) abruptly
- Can precipitate acute worsening in 30-50% within 7-10 days, potentially causing crisis
- ✅ Correct: Start 10mg, increase by 5mg every 3-5 days
❌ Not imaging the thymus
- Missing thymoma (10-20% of AChR-positive MG)
- ✅ Correct: CT/MRI thorax mandatory in all MG patients at diagnosis
❌ Not testing for MuSK antibodies in seronegative cases
- Missing 5-8% of MG cases with different treatment requirements
- ✅ Correct: Test AChR → if negative, test MuSK → if negative, test LRP4
❌ Prescribing aminoglycosides, fluoroquinolones, or beta-blockers
- Can precipitate myasthenic crisis
- ✅ Correct: Always screen medications; use alternative antibiotics (penicillins, cephalosporins)
❌ Waiting for hypoxia before intubating in crisis
- Hypoxia is late sign; delay risks cardiac arrest
- ✅ Correct: Intubate based on FVC less than 15mL/kg, NIF, declining trend, or bulbar weakness
❌ Recommending thymectomy for MuSK-positive MG
- No benefit (thymus not involved in pathogenesis)
- ✅ Correct: Thymectomy for thymoma (mandatory) or generalised AChR-positive MG age 18-65
❌ Stating pyridostigmine modifies disease course
- Purely symptomatic; does not alter disease progression
- ✅ Correct: Pyridostigmine provides symptomatic relief; immunosuppression is disease-modifying
❌ Confusing myasthenic crisis with cholinergic crisis
- Myasthenic: Under-treatment, weakness, tachycardia, sweating, dilated pupils
- Cholinergic: Over-treatment with AChE inhibitors, weakness plus secretions, miosis, bradycardia
- ✅ Correct: Temporarily stop pyridostigmine—improvement suggests cholinergic crisis
Clinical Pearls for Exams
-
Ice pack test: 80-90% sensitive for ocular MG with ptosis; immediate bedside test [12]
-
Steroid initiation: Start LOW and SLOW (10mg, increase 5mg per 3-5 days) to avoid worsening [1,13]
-
MuSK-MG triad: Poor pyridostigmine response + No thymectomy benefit + Excellent rituximab response [9]
-
Crisis intubation criteria: FVC less than 15mL/kg (don't wait for hypoxia)
-
MGTX trial: Proved thymectomy benefit in generalised AChR-positive MG age 18-65 [7]
-
Antibody sensitivity: AChR 85% (generalised), 50% (ocular); MuSK 5-8% (if AChR-negative)
-
SF-EMG: Most sensitive test (95-99%) but least specific; RNS more specific but less sensitive (50-60%)
-
Thymoma frequency: 10-20% of AChR-positive MG; mandatory thymectomy [6]
-
Crisis mortality: less than 5% with modern ICU care (down from 30-40% historically) [5]
-
Drug avoidance: Aminoglycosides, fluoroquinolones, beta-blockers, magnesium [14]
14. Patient Explanation (Layperson Level)
What is Myasthenia Gravis?
Myasthenia gravis is a condition where your immune system—which normally protects you from infections—mistakenly attacks the connections between your nerves and muscles. These connections are called neuromuscular junctions, and they allow your nerves to tell your muscles to move.
In myasthenia gravis, your immune system produces antibodies (proteins) that damage these connections. This means the signals from your nerves don't reach your muscles properly, causing muscle weakness.
The hallmark feature is that the weakness gets worse when you use your muscles repeatedly and improves after you rest. For example, your eyelids might droop more at the end of the day, or your voice might become weaker after talking for a while.
What causes it?
We don't fully understand why the immune system starts attacking the neuromuscular junctions. In many people, the thymus gland (a small organ in your chest that helps train your immune system) is abnormal—either enlarged or containing a benign or malignant tumour called a thymoma.
There is a genetic component, meaning it can run in families, but it's not directly inherited like some genetic conditions.
What are the symptoms?
Common symptoms include:
- Drooping eyelids (ptosis) and double vision (diplopia)—usually the first symptoms
- Difficulty speaking, chewing, or swallowing—your voice may sound nasal or weak
- Weakness in arms and legs—difficulty lifting objects, climbing stairs, or standing from a chair
- Breathing difficulties in severe cases
The weakness fluctuates—you might feel relatively normal in the morning but much weaker by evening. Symptoms improve with rest.
How is it diagnosed?
Your doctor will diagnose myasthenia gravis through:
- Blood tests looking for antibodies (proteins) that attack the neuromuscular junction
- Nerve tests (electromyography) showing how well your nerves and muscles communicate
- Ice pack test: Applying ice to a droopy eyelid for 2 minutes often improves the droop if you have myasthenia gravis
- CT or MRI scan of your chest to check for thymus gland abnormalities or tumours
How is it treated?
Treatment aims to improve muscle strength and reduce the immune system's attack. Options include:
-
Pyridostigmine: A medication that improves communication between nerves and muscles, reducing weakness. You take it 3-4 times daily.
-
Immunosuppression: Medications that calm your immune system, such as:
- Prednisolone (steroid)—started at a low dose and increased gradually
- Azathioprine or mycophenolate—steroid-sparing drugs that allow lower steroid doses
-
Thymectomy: Surgery to remove the thymus gland. This is recommended for most people with generalized myasthenia gravis under age 65, and is essential if you have a thymoma (tumour).
-
Emergency treatments: If you develop severe weakness affecting breathing (called myasthenic crisis), you may need:
- Plasma exchange (removing antibodies from blood)
- IVIG (infusion of antibodies from donors that help regulate your immune system)
- Intensive care with breathing support if needed
What is myasthenic crisis?
This is a medical emergency where the weakness becomes so severe that it affects your breathing muscles. Warning signs include:
- Severe difficulty breathing or shortness of breath
- Difficulty swallowing or talking
- Rapidly worsening weakness
If you experience these symptoms, call emergency services or go to hospital immediately.
What should I avoid?
Certain medications can worsen myasthenia gravis, including some antibiotics (gentamicin, ciprofloxacin), blood pressure medications (beta-blockers), and magnesium supplements. Always tell doctors, dentists, and pharmacists that you have myasthenia gravis before receiving any new medications.
What is the outlook?
With modern treatment, most people with myasthenia gravis can live normal or near-normal lives. About 20-30% of people achieve complete remission (no symptoms, no medications). The key is finding the right combination of treatments for you and working closely with your medical team.
The condition requires lifelong monitoring, but with appropriate treatment, the outlook is excellent.
15. References
-
Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: Executive summary. Neurology. 2016;87(4):419-425. doi:10.1212/WNL.0000000000002790
-
Gilhus NE. Myasthenia gravis-Pathophysiology, diagnosis, and treatment. Handb Clin Neurol. 2024;200:283-305. doi:10.1016/B978-0-12-823912-4.00026-8
-
Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14(10):1023-1036. doi:10.1016/S1474-4422(15)00145-3
-
Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat Rev Dis Primers. 2019;5(1):30. doi:10.1038/s41572-019-0079-y
-
Claytor B, Lowry TJ. Myasthenic crisis. Muscle Nerve. 2023;68(1):8-19. doi:10.1002/mus.27832
-
Romi F, Gilhus NE, Aarli JA. Myasthenia gravis: clinical, immunological, and therapeutic advances. Acta Neurol Scand. 2005;111(3):134-141. doi:10.1111/j.1600-0404.2005.00373.x
-
Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized Trial of Thymectomy in Myasthenia Gravis. N Engl J Med. 2016;375(6):511-522. doi:10.1056/NEJMoa1602489
-
Hehir MK, Silvestri NJ. Generalized Myasthenia Gravis: Classification, Clinical Presentation, Natural History, and Epidemiology. Neurol Clin. 2018;36(2):253-260. doi:10.1016/j.ncl.2018.01.002
-
Vesperinas-Castro A, Cortes-Vicente E. Rituximab treatment in myasthenia gravis. Front Neurol. 2023;14:1275533. doi:10.3389/fneur.2023.1275533
-
Zain A, Akram MS, Ashfaq F, et al. Comparative Analysis of Intravenous Immunoglobulins (IVIg) vs Plasmapheresis (PLEX) in the Management of Myasthenic Crisis. Cureus. 2024;16(9):e68895. doi:10.7759/cureus.68895
-
Bubuioc AM, Kudebayeva A, Turuspekova S, Lisnic V, Leone MA. The epidemiology of myasthenia gravis. J Med Life. 2021;14(1):7-16. doi:10.25122/jml-2020-0145
-
Sethi KD, Rivner MH, Swift TR. Ice pack test for myasthenia gravis: value and limitations. Neurology. 1987;37(8):1383-1385. doi:10.1212/wnl.37.8.1383
-
Mehndiratta MM, Pandey S, Kuntzer T. Acetylcholinesterase inhibitor treatment for myasthenia gravis. Cochrane Database Syst Rev. 2014;(10):CD006986. doi:10.1002/14651858.CD006986.pub3
-
Wittbrodt ET. Drugs and myasthenia gravis. An update. Arch Intern Med. 1997;157(4):399-408.
-
Gregersen PK, Kosoy R, Lee AT, et al. Risk for myasthenia gravis maps to a (151) Pro→Ala change in TNIP1 and to human leukocyte antigen-B*08. Ann Neurol. 2012;72(6):927-935. doi:10.1002/ana.23691
-
Jaretzki A 3rd, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. 2000;55(1):16-23. doi:10.1212/wnl.55.1.16
-
Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10(12):1098-1107. doi:10.1016/S1474-4422(11)70245-9
-
Association between Myasthenia Gravis and Smoking (Japan MG Registry 2021). Intern Med. 2025;64(24):3478-3485. doi:10.2169/internalmedicine.5310-25
-
Evoli A. Myasthenia gravis: new developments in research and treatment. Curr Opin Neurol. 2017;30(5):464-470. doi:10.1097/WCO.0000000000000473
-
Juel VC, Massey JM. Myasthenia gravis. Orphanet J Rare Dis. 2007;2:44. doi:10.1186/1750-1172-2-44
-
Palace J, Lashley D, Newsom-Davis J, et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain. 2007;130(Pt 6):1507-1515. doi:10.1093/brain/awm072
-
Spillane J, Hayward M, Hirsch NP, Taylor C, Kullmann DM, Howard RS. Thymectomy: role in the treatment of myasthenia gravis. J Neurol. 2013;260(7):1798-1801. doi:10.1007/s00415-013-6832-6
Last Updated: 2026-01-09
Frequently asked questions
Quick clarifications for common clinical and exam-facing questions.
When should I seek emergency care for myasthenia gravis?
Seek immediate emergency care if you experience any of the following warning signs: Respiratory muscle weakness (MG crisis), Bulbar symptoms (dysphagia, dysarthria), Rapid deterioration, FVC less than 1L or declining, Unable to count to 20 in one breath, Aspiration risk.
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Neuromuscular Junction Physiology
- Autoimmune Disorders - Overview
Differentials
Competing diagnoses and look-alikes to compare.
- Lambert-Eaton Myasthenic Syndrome
- Botulism
- Motor Neuron Disease
Consequences
Complications and downstream problems to keep in mind.
- Respiratory Failure
- Aspiration Pneumonia