Polyarteritis Nodosa
Polyarteritis nodosa (PAN) is a rare necrotizing vasculitis that primarily affects medium-sized muscular arteries, leadi... MRCP exam preparation.
What matters first
Polyarteritis nodosa (PAN) is a rare necrotizing vasculitis that primarily affects medium-sized muscular arteries, leadi... MRCP exam preparation.
Mesenteric ischaemia and bowel perforation
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Mesenteric ischaemia and bowel perforation
- Acute renal failure with renal infarction
- Severe peripheral neuropathy with muscle weakness
- Cardiac involvement with MI or heart failure
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- Microscopic Polyangiitis
- Granulomatosis with Polyangiitis
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Polyarteritis Nodosa
1. Overview
Polyarteritis nodosa (PAN) is a rare necrotizing vasculitis that primarily affects medium-sized muscular arteries, leading to multisystem involvement with significant morbidity and mortality if untreated. [1] First described by Kussmaul and Maier in 1866, PAN represents a distinct clinical entity within the systemic vasculitides, characterized by its ANCA-negative status and absence of glomerulonephritis—features that distinguish it from the ANCA-associated vasculitides. [2]
The disease manifests through transmural inflammation and fibrinoid necrosis of arterial walls, resulting in microaneurysm formation, thrombosis, and end-organ ischaemia. [3] PAN demonstrates a predilection for specific vascular beds including renal, gastrointestinal, peripheral nervous, and skin arteries, while notably sparing the pulmonary circulation. This selective involvement creates a characteristic clinical syndrome of constitutional symptoms, cutaneous manifestations (livedo reticularis, nodules), mononeuritis multiplex, renovascular hypertension, and abdominal pain. [4]
The association between PAN and hepatitis B virus (HBV) infection, first recognized in 1970, represents a unique paradigm in vasculitis pathogenesis, though its prevalence has declined substantially with widespread HBV vaccination. [5] Modern diagnostic approaches combine clinical assessment with characteristic angiographic findings (microaneurysms and stenoses) or histopathological confirmation. Treatment strategies have evolved from corticosteroid monotherapy to combination immunosuppression, with HBV-associated PAN requiring specific antiviral approaches and plasma exchange. [6] With contemporary management, 5-year survival exceeds 80%, representing a dramatic improvement from historical mortality rates of 90% at 5 years. [7]
2. Epidemiology
Incidence and Prevalence
Polyarteritis nodosa is a rare disease with an estimated annual incidence of 2-9 cases per million population in Western countries. [1] European epidemiological studies report incidences ranging from 4.4 to 9.7 per million per year, with variations likely reflecting differences in diagnostic criteria, HBV prevalence, and case ascertainment methods. [8] The prevalence of PAN has been estimated at approximately 30-31 cases per million population. [9]
Temporal trends demonstrate a significant decline in PAN incidence over recent decades, primarily attributable to widespread hepatitis B vaccination programs and improved HBV screening of blood products. [5] Studies from France showed a reduction in annual incidence from 7.7 per million in 1988-1989 to 3.1 per million in 2008-2009. [10] This decline parallels the decrease in HBV-associated PAN, which now accounts for less than 5-10% of cases in developed countries, compared to 30-40% in the pre-vaccination era. [11]
| Parameter | Value | Source |
|---|---|---|
| Annual incidence | 2-9 per million | [1,8] |
| Prevalence | 30-31 per million | [9] |
| HBV-associated PAN (current) | 5-10% | [11] |
| HBV-associated PAN (pre-vaccination) | 30-40% | [11] |
| 5-year survival (treated) | 80-90% | [7] |
| 5-year survival (untreated, historical) | 10-15% | [7] |
Demographics
PAN demonstrates a slight male predominance with a male-to-female ratio ranging from 1.5:1 to 2:1 across most studies. [1,8] The disease can occur at any age but shows peak incidence in the fifth and sixth decades of life, with a mean age at diagnosis of 45-55 years. [4] Paediatric PAN is rare but recognized, with distinct features including higher frequency of cutaneous and musculoskeletal involvement. [12]
Ethnic variations in incidence have been reported, with higher rates observed in populations with elevated HBV prevalence. Asian populations, particularly in regions with endemic hepatitis B, historically demonstrated higher PAN incidence. [5] However, this geographic pattern has attenuated with HBV vaccination programs. No consistent racial or genetic predisposition has been definitively established for non-HBV-associated PAN. [1]
Risk Factors
The most significant established risk factor for PAN is chronic hepatitis B infection, though this association has diminished in prevalence with vaccination. [5] HBV-associated PAN typically develops within 6 months of acute hepatitis B infection, during the period of active viral replication and immune complex formation. [11]
Additional proposed risk factors and associations include:
- Infectious agents: Hepatitis C virus (rare), HIV, parvovirus B19, cytomegalovirus (case reports, causality uncertain). [13]
- Medications: Minocycline, propylthiouracil, amphetamines (rare drug-induced vasculitis mimicking PAN). [14]
- Hairy cell leukaemia: Well-documented association with cutaneous and systemic PAN. [15]
- Familial Mediterranean fever: Increased vasculitis risk in some populations. [16]
Unlike ANCA-associated vasculitides, PAN shows no clear association with HLA haplotypes or specific genetic polymorphisms, suggesting environmental or infectious triggers predominate over genetic susceptibility. [1]
3. Aetiology and Pathophysiology
Aetiology
The aetiology of PAN remains incompletely understood, with the majority of cases classified as idiopathic. [1] Hepatitis B virus represents the only definitively established aetiological agent, accounting for 5-10% of contemporary PAN cases in developed countries. [11] The temporal relationship between acute HBV infection and vasculitis onset, combined with detection of viral antigens and immune complexes in vessel walls, provides strong aetiological evidence. [5]
Other proposed infectious triggers include hepatitis C (rare, with most HCV-associated vasculitis representing cryoglobulinaemic vasculitis rather than true PAN), parvovirus B19, and HIV, though these associations lack the robust evidence supporting HBV. [13] Drug-induced PAN-like vasculitis has been reported with minocycline, propylthiouracil, and amphetamines, though these may represent distinct pathological entities. [14]
The pathogenic mechanisms underlying idiopathic PAN remain speculative. Proposed hypotheses include:
- Unidentified infectious triggers with molecular mimicry
- Immune dysregulation with loss of tolerance to vascular antigens
- Complement activation through alternative pathway
- Endothelial cell activation and injury from circulating immune complexes
Pathophysiology
The pathophysiology of PAN centres on immune-mediated necrotizing inflammation of medium-sized muscular arteries, with distinct mechanisms for HBV-associated and idiopathic forms.
HBV-Associated PAN
In HBV-related disease, the pathogenic sequence involves:
-
Immune complex formation: During active HBV replication, hepatitis B surface antigen (HBsAg) combines with host antibodies to form circulating immune complexes. [5]
-
Vascular deposition: These immune complexes deposit in medium-vessel arterial walls, triggered by factors including turbulent blood flow, increased vascular permeability, and complement activation. [17]
-
Complement activation: Deposited immune complexes activate the classical complement pathway, generating anaphylatoxins (C3a, C5a) that recruit neutrophils and amplify inflammation. [17]
-
Neutrophil infiltration: Activated neutrophils infiltrate the arterial wall, releasing proteolytic enzymes, reactive oxygen species, and neutrophil extracellular traps (NETs) that cause direct endothelial and smooth muscle damage. [3]
-
Fibrinoid necrosis: Progressive transmural inflammation leads to fibrinoid necrosis—the pathological hallmark of PAN—characterized by vessel wall necrosis with fibrin deposition. [3]
Idiopathic PAN
In non-HBV-associated PAN, the pathophysiology is less clearly defined but appears to involve:
-
Innate immune activation: Possible triggers (unidentified pathogens, endogenous danger signals) activate toll-like receptors (TLRs) and inflammasomes, initiating inflammatory cascades. [18]
-
Endothelial activation: Pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) induce endothelial cell activation with upregulation of adhesion molecules (ICAM-1, VCAM-1, E-selectin). [18]
-
Leukocyte recruitment: Activated endothelium recruits neutrophils, monocytes, and T lymphocytes through sequential adhesion cascade (rolling, firm adhesion, transmigration). [3]
-
Transmural inflammation: Inflammatory cells penetrate the full thickness of the arterial wall (tunica intima, media, and adventitia), distinguishing PAN from more superficial vasculitides. [3]
-
Matrix degradation: Matrix metalloproteinases (MMPs) released by inflammatory cells degrade collagen and elastin, weakening arterial walls and predisposing to aneurysm formation. [19]
Vascular Consequences
The inflammatory process produces three principal vascular sequelae:
-
Microaneurysm formation: Destruction of the arterial media weakens vessel walls, leading to focal dilatation and microaneurysm formation at branch points where haemodynamic stress concentrates. [3] These appear as characteristic "beading" on angiography.
-
Thrombosis and stenosis: Endothelial damage, flow disturbance, and inflammatory thrombosis cause luminal narrowing and occlusion, resulting in distal ischaemia. [4]
-
End-organ infarction: Critical stenosis or thrombotic occlusion produces tissue infarction in supplied territories, manifesting clinically as renal infarcts, bowel ischaemia, myocardial infarction, or stroke. [4]
Organ-Specific Manifestations
The predilection for specific vascular beds reflects:
-
Vessel size preference: Medium muscular arteries (100-400 μm diameter) are selectively affected; capillaries, venules, and large elastic arteries are characteristically spared. [2]
-
Anatomical distribution: Renal arcuate and interlobar arteries, mesenteric and hepatic arterial branches, vasa nervorum of peripheral nerves, and dermal arterioles demonstrate preferential involvement. [4]
-
Pulmonary sparing: The pulmonary circulation is typically spared, a key distinguishing feature from ANCA-associated vasculitis and a criterion in Chapel Hill classification. [2] The mechanism underlying pulmonary sparing remains unclear but may relate to unique pulmonary endothelial properties or haemodynamic factors.
Temporal Evolution
PAN demonstrates segmental and focal involvement with lesions at different evolutionary stages:
- Acute phase: Active neutrophilic inflammation with fibrinoid necrosis
- Subacute phase: Mixed inflammatory infiltrate with eosinophils, lymphocytes, and monocytes
- Chronic phase: Fibrosis, intimal proliferation, and organized thrombi with recanalization
This asynchronous evolution produces coexisting acute and chronic lesions—a pathological feature distinguishing PAN from vasculitides with more uniform temporal distribution. [3]
The absence of anti-neutrophil cytoplasmic antibodies (ANCA) and immune complex deposits in many idiopathic cases suggests pathogenic mechanisms distinct from both ANCA-associated vasculitis and classic immune complex diseases. [1] Current research explores roles for cellular immunity, innate immune dysregulation, and alternative complement activation, though definitive mechanisms remain elusive.
4. Clinical Presentation
Polyarteritis nodosa typically presents with a subacute, progressive course over weeks to months, though acute presentations can occur, particularly with life-threatening complications. [4] The clinical manifestations reflect multisystem involvement with varying combinations of constitutional symptoms and organ-specific features.
Constitutional Symptoms
Systemic features are nearly universal in PAN and often represent the initial manifestations:
- Fever: Present in 50-60% of patients, typically low-grade to moderate (38-39°C), though high spiking fevers can occur with extensive disease or complications. [4]
- Weight loss: Involuntary weight loss exceeding 4-5 kg occurs in 40-60% of patients, reflecting systemic inflammation and increased metabolic demands. [4]
- Malaise and fatigue: Profound fatigue disproportionate to activity level affects 70-80% of patients. [20]
- Myalgias and arthralgias: Muscle and joint pain without frank arthritis occurs in 50-70%, predominantly affecting large muscle groups and proximal joints. [4]
These non-specific features often lead to diagnostic delays, with patients undergoing extensive investigation for occult malignancy, infection, or other systemic diseases before PAN diagnosis. [20]
Cutaneous Manifestations (50-60%)
Skin involvement occurs in approximately 50-60% of patients and may provide visible diagnostic clues: [4]
Livedo reticularis (30-50%): A mottled, reticular, purplish discoloration of the skin, most prominent on lower extremities and trunk, results from inflammatory involvement of dermal and subcutaneous arterioles causing sluggish blood flow. [21] Unlike livedo racemosa or physiological livedo, PAN-associated livedo reticularis is persistent, irregular, and may be associated with ulceration. [21]
Subcutaneous nodules (15-20%): Tender, palpable nodules along the course of superficial arteries, particularly on lower extremities, represent inflamed arterial segments or small aneurysms. [4] These nodules may wax and wane with disease activity and can be biopsied for histological confirmation.
Skin ulcers (10-20%): Painful ulcerations, typically on distal extremities, result from cutaneous infarction secondary to arterial occlusion. [21] These ulcers often have irregular borders, punched-out appearance, and poor healing without immunosuppressive treatment.
Digital ischaemia: Finger and toe ischaemia, manifesting as painful digital infarcts, gangrene, or splinter haemorrhages, occurs in 5-10% of patients. [4]
Other cutaneous features: Purpura, ecchymoses, urticaria, and erythematous macules/papules are less specific but may occur. [21]
Neurological Manifestations (50-75%)
Peripheral nervous system involvement represents one of the most characteristic features of PAN: [4]
Mononeuritis multiplex (50-70%): The pathognomonic neurological manifestation of PAN, mononeuritis multiplex results from ischaemic injury to vasa nervorum supplying peripheral nerves. [22] Clinical features include:
- Asymmetric, multifocal sensory and motor deficits
- Acute or subacute onset of pain, paresthesias, and weakness in the distribution of individual nerves
- Common patterns include foot drop (peroneal nerve), wrist drop (radial nerve), and sensory loss in specific nerve distributions
- Stepwise accumulation of deficits over days to weeks as additional nerves become affected
- EMG/nerve conduction studies demonstrate axonal neuropathy with asymmetric, multifocal pattern [22]
Peripheral polyneuropathy (20-30%): Some patients develop symmetric distal sensorimotor polyneuropathy, likely reflecting confluence of multiple mononeuropathies or more diffuse small vessel involvement. [22]
Central nervous system involvement (5-10%): CNS manifestations are less common but serious:
- Stroke (ischaemic or haemorrhagic) from cerebral arteritis
- Seizures (focal or generalized)
- Encephalopathy or altered mental status
- Cranial neuropathies (rare)
- Headache (non-specific but may indicate CNS vasculitis) [4]
Renal Manifestations (70-80%)
Renal involvement is one of the most common and prognostically significant features of PAN: [4]
Renovascular hypertension (50-70%): Hypertension, often severe and difficult to control, results from renal ischaemia activating the renin-angiotensin-aldosterone system. [23] Acute-onset hypertension in a previously normotensive middle-aged adult should raise suspicion for PAN.
Renal infarction (30-50%): Segmental or global renal infarction from renal artery branch occlusion manifests with:
- Acute flank pain
- Haematuria (microscopic or gross)
- Elevated lactate dehydrogenase (LDH)
- Acute kidney injury [23]
Progressive renal impairment (20-40%): Cumulative ischaemic injury leads to progressive chronic kidney disease. Serial measurements show rising creatinine and declining GFR. [23]
Importantly, glomerulonephritis is absent in classic PAN—a key distinguishing feature from ANCA-associated vasculitides. [2] Urinalysis typically shows bland sediment without significant proteinuria, red cell casts, or dysmorphic erythrocytes. The presence of active glomerulonephritis should prompt reconsideration of the diagnosis in favor of microscopic polyangiitis or other glomerulonephritides.
Renal artery aneurysms: Multiple renal microaneurysms (0.5-1 cm) are pathognomonic when visualized on angiography, appearing as characteristic beading or rosary pattern. [23]
Gastrointestinal Manifestations (40-60%)
GI involvement carries significant morbidity and mortality risk: [4]
Abdominal pain (40-50%): The most common GI symptom, abdominal pain results from mesenteric arteritis causing intestinal ischaemia. Pain is typically:
- Postprandial (mesenteric angina)
- Colicky or cramping in character
- Diffuse or periumbilical
- Associated with food aversion and weight loss [24]
Mesenteric ischaemia and infarction (15-20%): Acute mesenteric ischaemia represents a surgical emergency:
- Severe, acute abdominal pain out of proportion to physical findings
- Nausea, vomiting, diarrhoea (sometimes bloody)
- Peritoneal signs if perforation occurs
- Elevated lactate, leukocytosis
- CT angiography shows arterial occlusion or characteristic aneurysms [24]
Gastrointestinal bleeding (10-15%): Manifests as haematemesis, melena, or haematochezia from mucosal ischaemia/ulceration. [24]
Bowel perforation (5-10%): Transmural infarction leads to perforation with peritonitis, requiring emergency surgical intervention. [24]
Hepatic involvement: Hepatic artery vasculitis can cause hepatic infarction, liver dysfunction, or present as right upper quadrant pain. [4]
Cholecystitis: Acalculous cholecystitis from cystic artery involvement is described. [4]
Cardiac Manifestations (30-50%)
Cardiac involvement significantly impacts prognosis: [4]
Coronary arteritis (10-20%): Inflammation of coronary arteries causes:
- Acute myocardial infarction (may occur in young patients without traditional risk factors)
- Angina pectoris
- Heart failure from ischaemic cardiomyopathy
- Coronary aneurysms (visible on CT coronary angiography) [25]
Pericarditis (10-15%): Presents with chest pain, pericardial friction rub, and ECG changes (diffuse ST elevation, PR depression). Pericardial effusion may develop. [25]
Heart failure (10-15%): Results from coronary ischaemia, hypertension, or direct myocardial involvement. [25]
Arrhythmias: Atrial and ventricular arrhythmias can occur from ischaemic or inflammatory myocardial injury. [25]
Valvular disease: Rare reports of valvulitis, though not a characteristic feature. [4]
Genitourinary Manifestations (15-25%)
Testicular involvement (10-15%): Orchitis from testicular artery vasculitis causes:
- Acute testicular pain and swelling
- Mimics testicular torsion or epididymo-orchitis
- Ultrasound may show heterogeneous echotexture and increased vascularity
- Testicular infarction can occur
- Orchidectomy specimens provide excellent tissue for histological diagnosis [26]
Ovarian involvement (rare): Ovarian infarction or haemorrhage reported in women. [4]
Bladder involvement (rare): Haematuria from bladder arteritis. [4]
Musculoskeletal Manifestations (40-60%)
Myalgias (50-60%): Diffuse muscle pain and tenderness, particularly affecting proximal muscles. [4]
Arthralgias (40-50%): Joint pain without objective synovitis, typically affecting large joints (knees, ankles, shoulders). [4]
Arthritis (10-20%): True inflammatory arthritis is less common but can occur as non-erosive polyarthritis. [20]
Muscle infarction (rare): Acute, painful muscle swelling from intramuscular arteritis and infarction. [4]
Other Organ Systems
Ophthalmological (10-15%):
- Retinal vasculitis with cotton-wool spots
- Choroidal infarction
- Anterior uveitis
- Optic neuropathy
- Visual loss from central retinal artery occlusion [4]
ENT (rare):
- Facial pain
- Hearing loss
- Parotid swelling
- Important: Destructive upper airway disease is absent in PAN and its presence suggests granulomatosis with polyangiitis. [2]
Breast (rare): Breast infarction from mammary artery involvement (case reports). [4]
Clinical Presentation Patterns
PAN classically presents in one of three patterns:
-
Subacute multisystem disease (most common): Gradual onset over weeks to months with accumulating constitutional symptoms and multiorgan involvement.
-
Acute catastrophic presentation: Rapid onset with life-threatening complications (mesenteric infarction, MI, stroke) requiring urgent intervention.
-
Cutaneous-limited PAN: Predominantly or exclusively cutaneous manifestations without significant systemic involvement (controversial whether this represents true PAN or a distinct entity).
5. Differential Diagnosis
The differential diagnosis of PAN is broad, reflecting its protean multisystem manifestations. Distinguishing PAN from other systemic vasculitides and mimicking conditions is essential for appropriate management.
Primary Differential: ANCA-Associated Vasculitides
The ANCA-associated vasculitides (AAV)—microscopic polyangiitis (MPA), granulomatosis with polyangiitis (GPA), and eosinophilic granulomatosis with polyangiitis (EGPA)—represent the primary diagnostic challenge.
| Feature | PAN | Microscopic Polyangiitis | Granulomatosis with Polyangiitis | EGPA |
|---|---|---|---|---|
| ANCA | Negative | Positive (MPO-ANCA 60%) | Positive (PR3-ANCA 80%) | Positive (40-60%) |
| Vessel size | Medium arteries | Small vessels (capillaries) | Small-medium vessels | Small-medium vessels |
| Glomerulonephritis | Absent | Prominent (pauci-immune) | Common (pauci-immune) | Less common |
| Pulmonary involvement | Absent | Common (alveolar haemorrhage) | Common (nodules, cavitation) | Prominent (asthma, infiltrates) |
| Granulomas | Absent | Absent | Present (necrotizing) | Present (eosinophilic) |
| Eosinophilia | Absent | Absent | Absent | Prominent (> 10%) |
| Upper airway disease | Absent | Absent | Characteristic (sinusitis, saddle nose) | Common (rhinosinusitis, polyps) |
| Mononeuritis multiplex | Characteristic | Possible | Possible | Common |
| Aneurysms on angiography | Characteristic | Absent | Rare | Rare |
| HBV association | 5-10% | Rare | Rare | Rare |
Microscopic polyangiitis (MPA) [27]: The closest differential to PAN, MPA differs by:
- Small vessel (capillary) involvement rather than medium arteries
- Necrotizing glomerulonephritis (nearly universal)
- Pulmonary capillaritis with alveolar haemorrhage (common)
- MPO-ANCA positivity (60-80%)
- Absence of microaneurysms on angiography
Granulomatosis with polyangiitis (GPA, formerly Wegener's) [28]:
- Upper respiratory tract involvement (sinusitis, nasal crusting, saddle nose deformity)
- Pulmonary nodules or cavitary lesions
- Necrotizing granulomatous inflammation on biopsy
- PR3-ANCA positivity (80-90%)
- Can have glomerulonephritis
Eosinophilic granulomatosis with polyangiitis (EGPA, formerly Churg-Strauss) [29]:
- Adult-onset asthma (nearly universal)
- Peripheral eosinophilia (> 1000/μL)
- Pulmonary infiltrates
- Paranasal sinus disease
- ANCA-positive in 40-60% (typically MPO-ANCA)
Other Vasculitides
Kawasaki disease [30]: Primarily pediatric medium-vessel vasculitis that can resemble PAN:
- Age: Predominantly children less than 5 years (vs adults in PAN)
- Diagnostic criteria: Fever > 5 days plus conjunctivitis, oral changes, rash, extremity changes, cervical lymphadenopathy
- Coronary artery aneurysms: Characteristic complication
- Self-limited course (vs chronic in PAN)
- Treatment: IVIG and aspirin (vs immunosuppression in PAN)
Cryoglobulinaemic vasculitis [31]: Associated with hepatitis C (vs hepatitis B in PAN):
- Palpable purpura (prominent)
- Arthralgias/arthritis
- Membranoproliferative glomerulonephritis
- Low C4 complement levels (vs normal in PAN)
- Positive cryoglobulins
- HCV antibodies and RNA positive
IgA vasculitis (Henoch-Schönlein purpura) [32]: Small vessel vasculitis with:
- Palpable purpura (buttocks and lower extremities)
- Abdominal pain and GI bleeding
- IgA nephropathy
- Arthralgias
- More common in children
- IgA deposition on biopsy
Behçet's disease [33]: Vasculitis affecting arteries and veins:
- Recurrent oral and genital ulcers
- Uveitis
- Pathergy test positive
- Venous thrombosis
- Geographic predilection (Mediterranean, Middle East, East Asia)
Takayasu arteritis [34]: Large-vessel vasculitis:
- Young women (typically less than 40 years)
- Aorta and major branches involved
- Limb claudication, pulse discrepancies
- Vascular stenoses (not aneurysms) on imaging
- Elevated inflammatory markers
Non-Vasculitic Mimics
Infective endocarditis [35]: Can mimic PAN with:
- Fever, weight loss, constitutional symptoms
- Multisystem embolic phenomena
- Renal involvement (embolic infarcts, glomerulonephritis)
- Cutaneous manifestations (Osler nodes, Janeway lesions, splinter haemorrhages)
- Distinguishing features: Heart murmur, positive blood cultures, vegetations on echocardiography, response to antibiotics
Atrial myxoma: Cardiac tumor causing embolic phenomena:
- Constitutional symptoms (fever, weight loss)
- Multiorgan embolic infarcts
- Elevated inflammatory markers
- Distinguishing features: Cardiac mass on echocardiography, diastolic "plop" sound, removal curative
Cholesterol emboli syndrome [36]: Following arterial instrumentation or spontaneous:
- Livedo reticularis (livedo racemosa pattern)
- Renal failure
- GI symptoms
- Digital ischaemia
- Distinguishing features: Recent vascular procedure, eosinophilia, eosinophiluria, hypocomplementaemia, cholesterol crystals in retinal vessels
Antiphospholipid syndrome [37]: Autoimmune thrombophilia:
- Recurrent arterial/venous thrombosis
- Pregnancy morbidity
- Livedo reticularis
- Renal thrombotic microangiopathy
- Distinguishing features: Positive antiphospholipid antibodies (lupus anticoagulant, anticardiolipin, anti-β2-glycoprotein I), thrombotic rather than inflammatory vascular occlusion
Malignancy: Occult malignancy can mimic PAN:
- Constitutional symptoms (fever, weight loss)
- Elevated inflammatory markers
- Multiorgan dysfunction
- Paraneoplastic vasculitis (rare)
- Distinguishing features: Age-appropriate cancer screening, imaging for occult malignancy, biopsy confirmation
Drug-induced vasculitis [14]: Particularly cocaine, amphetamines, minocycline:
- Temporal relationship to drug exposure
- ANCA may be positive (especially with propylthiouracil, hydralazine, minocycline)
- Resolution with drug cessation
Fibromuscular dysplasia [38]: Non-inflammatory arteriopathy:
- Renal artery stenosis with hypertension
- "String of beads" appearance on angiography (can mimic microaneurysms)
- Distinguishing features: Younger women, no systemic symptoms, normal inflammatory markers, absence of other organ involvement
Segmental arterial mediolysis: Rare non-inflammatory arteriopathy:
- Abdominal pain from mesenteric involvement
- Aneurysms and dissections
- Distinguishing features: Absence of inflammation on histology, no systemic symptoms
6. Investigations
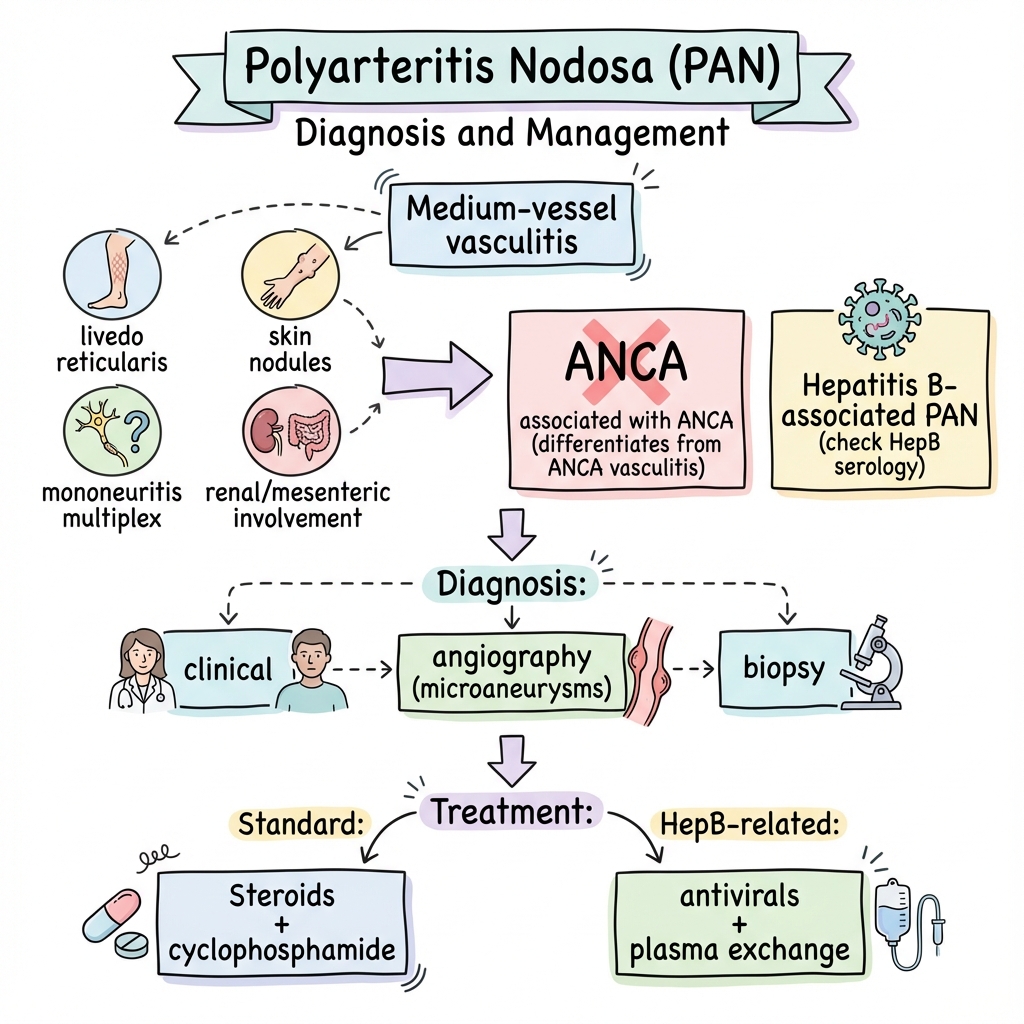
Diagnosis of PAN requires integration of clinical features, laboratory findings, imaging studies, and often histopathological confirmation, as no single test is pathognomonic. [1]
Laboratory Investigations
Essential First-Line Tests
Complete blood count (CBC):
- Normocytic anaemia of chronic disease (common)
- Leukocytosis with neutrophilia during active disease
- Thrombocytosis (reactive)
- Normal eosinophil count (elevated eosinophils suggest EGPA) [4]
Inflammatory markers:
- ESR: Typically markedly elevated (often > 50 mm/hr, frequently > 100 mm/hr) [4]
- CRP: Elevated (usually > 50 mg/L), correlates with disease activity [4]
- Serial measurements useful for monitoring treatment response
Renal function:
- Creatinine and estimated GFR: May show acute or chronic kidney injury
- Blood urea nitrogen: Elevated with renal involvement [23]
Urinalysis:
- Characteristically bland sediment (key distinguishing feature from ANCA vasculitis)
- Possible mild proteinuria (less than 1 g/24hr)
- Microscopic haematuria may occur with renal infarction
- Absence of red cell casts, dysmorphic erythrocytes, or significant proteinuria [2,23]
Liver function tests:
- Transaminases may be elevated with hepatic involvement
- Alkaline phosphatase elevation possible
- Hypoalbuminaemia (acute phase response) [4]
Critical Diagnostic Tests
Anti-neutrophil cytoplasmic antibodies (ANCA): [2]
- Must be negative for PAN diagnosis (Chapel Hill criteria)
- Both cytoplasmic (c-ANCA/PR3) and perinuclear (p-ANCA/MPO) patterns negative
- ANCA positivity mandates reconsideration of diagnosis toward ANCA-associated vasculitis
- Testing should include both indirect immunofluorescence and antigen-specific ELISA (PR3, MPO)
Hepatitis B serology: [5,11]
- Mandatory in all suspected PAN cases
- HBsAg (hepatitis B surface antigen): Positive in active HBV infection
- Anti-HBc (hepatitis B core antibody): Indicates past or current infection
- HBV DNA (viral load): Quantifies viral replication
- 5-10% of PAN cases are HBV-associated in developed countries
- Positive serology alters treatment approach (antivirals, plasma exchange)
Hepatitis C serology:
- Anti-HCV antibodies and HCV RNA
- Distinguishes cryoglobulinaemic vasculitis (HCV-associated) from true PAN [31]
HIV serology:
- HIV infection rarely associated with PAN-like vasculitis
- Necessary for comprehensive workup [13]
Additional Laboratory Tests
Complement levels:
- C3, C4 typically normal in PAN (distinguishes from cryoglobulinaemia, SLE)
- Low complement suggests alternative diagnosis [1]
Antinuclear antibodies (ANA):
- Usually negative or low titre
- High-titre positivity suggests overlap syndrome or SLE [1]
Cryoglobulins:
- Should be negative in PAN
- Positive cryoglobulins indicate cryoglobulinaemic vasculitis (often HCV-associated) [31]
Rheumatoid factor:
- May be positive in low titres (non-specific)
- High levels suggest rheumatoid vasculitis or cryoglobulinaemia [1]
Imaging Studies
Angiography (Gold Standard Imaging)
Conventional angiography or CT/MR angiography demonstrating characteristic vascular abnormalities provides strong diagnostic evidence for PAN. [39]
Findings: [39]
- Microaneurysms: Multiple small aneurysms (1-5 mm) at arterial branch points, creating characteristic "beading" or "rosary" appearance
- Arterial stenoses: Focal narrowing or occlusion of medium-sized arteries
- Irregular arterial contour: Alternating stenoses and dilations
- Distribution: Renal (most common), hepatic, mesenteric, and occasionally coronary arteries
Modalities:
- Conventional catheter angiography: Gold standard with highest resolution, allows sampling for angioplasty if indicated
- CT angiography (CTA): Non-invasive, excellent for abdominal vessels, identifies complications (infarcts, perforation)
- MR angiography (MRA): Alternative to CTA, avoids radiation and iodinated contrast
- Renal angiography: Most frequently abnormal, should be performed if PAN suspected and diagnosis uncertain
Sensitivity and Specificity: [39]
- Microaneurysms highly specific (> 90%) when present in appropriate clinical context
- Sensitivity ~70-80% (not all patients develop aneurysms, particularly early disease)
- False positives possible with fibromuscular dysplasia, segmental arterial mediolysis, mycotic aneurysms
Limitations:
- Normal angiography does not exclude PAN (particularly early disease)
- Cannot visualize vessels less than 100 μm (requires biopsy for small vessel involvement)
- Atherosclerotic disease may complicate interpretation in elderly
Cross-Sectional Imaging
CT abdomen/pelvis with contrast: [24]
- Identifies complications: bowel infarction, perforation, renal infarcts, splenic infarcts
- May show thickened bowel wall with mesenteric ischaemia
- Hepatic or renal hypodense areas suggesting infarction
- Free fluid or air indicating perforation
Magnetic resonance imaging (MRI):
- MR angiography for vessel assessment
- Brain MRI if CNS symptoms (ischaemic or haemorrhagic stroke)
- Cardiac MRI may show myocardial inflammation or infarction [25]
Chest imaging:
- Chest X-ray: Typically normal (pulmonary sparing is characteristic)
- Pulmonary infiltrates, nodules, or cavitation suggest alternative diagnosis (ANCA vasculitis)
Echocardiography: [25]
- Assess for pericardial effusion, wall motion abnormalities (ischaemia), valvular disease
- Rule out endocarditis (important differential)
Ultrasound:
- Testicular ultrasound if orchitis: heterogeneous echotexture, increased or decreased vascularity [26]
- Renal ultrasound: May show renal infarcts (though less sensitive than CT/MRI)
Neurophysiological Studies
Electromyography (EMG) and nerve conduction studies (NCS): [22]
- Essential for characterizing peripheral neuropathy
- Findings in mononeuritis multiplex:
- Asymmetric, multifocal axonal neuropathy
- Reduced amplitudes (motor and sensory)
- Normal or mildly reduced conduction velocities (axonal, not demyelinating)
- Absent responses in severely affected nerves
- Sequential testing demonstrates progression involving additional nerves
- Guides selection of nerve for biopsy (affected but not completely denervated)
Histopathology
Tissue biopsy provides definitive diagnosis but is not always necessary if clinical and angiographic features are characteristic. [1,3]
Biopsy Sites and Yield
| Biopsy Site | Diagnostic Yield | Indications | Risks |
|---|---|---|---|
| Sural nerve/muscle | 70-80% | Mononeuritis multiplex | Permanent sensory deficit |
| Skin | 40-60% | Nodules or ulcers | Low risk |
| Testis | 80-90% | Orchitis | Requires orchidectomy |
| Kidney | 30-50% | Renal involvement | Bleeding risk |
| Liver | Variable | Hepatic involvement | Significant bleeding risk |
Sural nerve and gastrocnemius muscle biopsy: [22]
- Combined nerve-muscle biopsy increases yield (70-80%)
- Demonstrates vasculitis of vasa nervorum
- Muscle biopsy alone lower yield (~50%)
- Histology: Acute inflammation with fibrinoid necrosis, or chronic changes with fibrosis and recanalization
Skin biopsy: [21]
- Sample subcutaneous nodule or edge of ulcer
- Include deep dermis and subcutaneous fat (medium vessels)
- Histology: Necrotizing vasculitis of small and medium arterioles
Testicular biopsy: [26]
- High diagnostic yield if orchitis present
- Usually requires orchidectomy (testicular infarction often present)
Renal biopsy: [23]
- Lower yield (30-50%) as arcuate arteries may not be sampled
- Risk of bleeding from vascular disease
- Critical: Absence of glomerulonephritis distinguishes PAN from ANCA vasculitis
- May show ischaemic changes, arteritis if sampled
Histological Features
Acute phase: [3]
- Transmural necrotizing inflammation
- Fibrinoid necrosis of vessel wall
- Neutrophilic infiltration with nuclear debris
- Disruption of internal elastic lamina
- Thrombosis with fibrin deposition
Subacute phase:
- Mixed inflammatory infiltrate (neutrophils, lymphocytes, macrophages, eosinophils)
- Intimal proliferation
- Continuing vessel wall necrosis
Chronic phase:
- Fibrotic thickening of vessel wall
- Adventitial fibrosis
- Organized thrombi with recanalization
- Intimal proliferation with luminal narrowing
Key features:
- Transmural inflammation (full thickness of arterial wall)
- Segmental involvement (skip lesions along vessel)
- Asynchronous lesions (coexisting acute, subacute, and chronic stages)
- Medium artery preference (100-400 μm diameter)
- Absence of granulomas (distinguishes from GPA, EGPA)
Diagnostic Criteria and Classification
Chapel Hill Consensus Conference (CHCC) 2012 Definition [2]
PAN is defined as:
"Necrotizing inflammation of medium or small arteries without glomerulonephritis or vasculitis in arterioles, capillaries, or venules, and not associated with ANCA."
Key definitional elements:
- Medium or small arteries affected
- Necrotizing inflammation
- Absence of glomerulonephritis
- Absence of arteriole, capillary, or venule vasculitis
- ANCA-negative
American College of Rheumatology (ACR) 1990 Classification Criteria [40]
Developed for classification (not diagnosis), requires ≥3 of 10 criteria:
- Weight loss ≥4 kg (not due to other cause)
- Livedo reticularis
- Testicular pain or tenderness
- Myalgias, weakness, or leg tenderness
- Mononeuropathy or polyneuropathy
- Diastolic BP > 90 mmHg (new-onset or worsening)
- Elevated BUN (> 40 mg/dL) or creatinine (> 1.5 mg/dL) not due to dehydration
- Hepatitis B virus (HBsAg or anti-HBs antibody)
- Arteriographic abnormality (aneurysms or occlusions of visceral arteries)
- Biopsy showing granulocytes in artery wall
Performance: Sensitivity 82%, specificity 87% for distinguishing PAN from other vasculitides [40]
Limitations: Developed pre-CHCC definition; does not require ANCA negativity; includes cutaneous-only features
Diagnostic Algorithm
- Clinical suspicion: Multisystem disease with constitutional symptoms, peripheral neuropathy, renal/GI involvement
- Laboratory screening: CBC, CRP/ESR, renal function, urinalysis, ANCA (must be negative), HBV serology
- Imaging: CT/MR angiography of renal and mesenteric vessels
- Biopsy (if diagnosis uncertain): Combined sural nerve/muscle biopsy or affected organ
- Classification: Apply CHCC definition and ACR criteria
Definitive diagnosis requires either:
- Characteristic angiographic findings (microaneurysms) + appropriate clinical features + ANCA-negative + HBV testing, OR
- Histological confirmation + appropriate clinical features + ANCA-negative + HBV testing
7. Classification and Prognostic Scoring
Five Factor Score (FFS)
The Five Factor Score (FFS), originally developed in 1996 and revised in 2009 (FFS-R), predicts mortality in PAN and provides prognostic stratification to guide treatment intensity. [41]
FFS (2009 Revised) for PAN [41]
Points assigned for presence of each factor:
| Factor | Points |
|---|---|
| Age > 65 years | +1 |
| Cardiac symptoms (heart failure, MI, pericarditis) | +1 |
| Gastrointestinal involvement (bleeding, perforation, infarction, pancreatitis) | +2 |
| Renal insufficiency (creatinine > 150 μmol/L or 1.7 mg/dL) | +1 |
Note: The original FFS included "absence of ENT manifestations" as a poor prognostic factor, but this was removed in revised FFS as ENT involvement is not typical of PAN (suggests GPA instead).
Risk stratification:
- FFS = 0: Low risk (5-year mortality ~10%)
- FFS = 1: Moderate risk (5-year mortality ~25%)
- FFS ≥2: High risk (5-year mortality ~40-50% without intensive treatment)
Clinical application:
- FFS = 0: Consider corticosteroid monotherapy
- FFS ≥1: Combination therapy with corticosteroids + cyclophosphamide recommended [41]
Disease Activity Indices
Birmingham Vasculitis Activity Score (BVAS): [42]
- Standardized assessment of disease activity across multiple organ systems
- Scores new or worsening manifestations in preceding 4 weeks
- Range 0-63 points
- Used in clinical trials and can guide treatment adjustments
- BVAS v3 is current version
Limitations: BVAS was developed primarily for ANCA-associated vasculitis; application to PAN is less validated but still useful for monitoring.
8. Management
Management of PAN has evolved substantially over recent decades, with modern immunosuppressive regimens achieving dramatic improvements in survival and morbidity. [6] Treatment approach depends on disease severity, presence of HBV association, and prognostic factors.
General Principles
- Early aggressive treatment for severe disease (FFS ≥1) to prevent irreversible organ damage
- Distinction between HBV-associated and non-HBV PAN as treatment differs fundamentally
- Induction therapy to achieve remission followed by maintenance therapy to prevent relapse
- Treat-to-target approach with regular disease activity assessment
- Management of complications and comorbidities (hypertension, infections, metabolic effects of corticosteroids)
Non-HBV-Associated PAN
Mild Disease (FFS = 0, No Organ-Threatening Manifestations)
Corticosteroid monotherapy: [6]
- Prednisolone: 1 mg/kg/day (maximum 60-80 mg/day) orally
- Continue high-dose for 4 weeks or until disease control
- Taper schedule:
- Reduce to 0.5 mg/kg/day over weeks 4-8
- Reduce to 10-15 mg/day by 3-6 months
- Slow taper to 5-7.5 mg/day by 12 months
- Attempt withdrawal at 18-24 months if sustained remission
- Response: 60-70% achieve remission with corticosteroids alone [6]
Alternative to prednisolone:
- Methylprednisolone: 0.8 mg/kg/day (equivalent dosing)
Monitoring:
- Clinical assessment every 2-4 weeks initially
- CRP/ESR every 2-4 weeks
- Adjust based on disease activity (BVAS)
Moderate-Severe Disease (FFS ≥1, Organ-Threatening Manifestations)
Combination therapy with corticosteroids + cyclophosphamide: [6,43]
Corticosteroid regimen:
Option 1 - Oral prednisolone:
- Prednisolone 1 mg/kg/day (max 60-80 mg/day)
- Taper as for mild disease once remission achieved
Option 2 - IV pulse methylprednisolone (for severe/life-threatening disease):
- Methylprednisolone 500-1000 mg IV daily for 3 days
- Followed by oral prednisolone 1 mg/kg/day
- Higher initial drug levels, potentially faster disease control
- Consider for severe renal involvement, mesenteric ischaemia, cardiac involvement, or CNS vasculitis
Cyclophosphamide regimen:
Option 1 - IV pulse cyclophosphamide (preferred):
- Dose: 15 mg/kg (maximum 1200 mg) IV every 2 weeks for 3 doses, then every 3 weeks
- Total induction duration: 3-6 months (typically 6 pulses)
- Dose adjustments:
- "Age > 60 years: Reduce dose by 25% (12.5 mg/kg)"
- "GFR 30-50 mL/min: Reduce dose by 25%"
- "GFR less than 30 mL/min: Reduce dose by 50% or avoid"
- Monitoring: FBC before each dose; delay if WBC less than 4.0 or neutrophils less than 2.0
Option 2 - Oral cyclophosphamide:
- Dose: 2 mg/kg/day orally (maximum 200 mg/day)
- Duration: 3-6 months
- Dose adjustments: As for IV route
- May have higher cumulative dose and bladder toxicity risk vs IV route
Evidence base:
- No randomized trials specifically in PAN (most evidence extrapolated from ANCA vasculitis trials)
- Observational data shows remission rates 80-90% with combination therapy [6]
- Reduced relapse rates compared to corticosteroid monotherapy
Supportive measures:
-
Pneumocystis jirovecii prophylaxis: [44]
- Trimethoprim-sulfamethoxazole 80/400 mg (single strength) daily or 160/800 mg (double strength) three times weekly
- "Alternative: Atovaquone 1500 mg daily or dapsone 100 mg daily (check G6PD first)"
- Continue throughout cyclophosphamide treatment and for 3-6 months after
-
GnRH agonists for fertility preservation: [45]
- Consider in women of childbearing age receiving cyclophosphamide
- Goserelin or leuprolide during cyclophosphamide treatment
- Does not guarantee fertility preservation but may reduce ovarian damage
-
Bladder protection:
- Aggressive hydration (2-3 L fluid daily)
- Mesna not routinely required with IV pulse cyclophosphamide
- Urinalysis periodically to screen for hemorrhagic cystitis
-
Bone protection: [46]
- Calcium 1000-1500 mg daily + Vitamin D 800-1000 IU daily
- Bisphosphonate (alendronate 70 mg weekly or risedronate 35 mg weekly) for patients on ≥7.5 mg prednisolone expected for ≥3 months
- DEXA scan at baseline and 1-2 years
-
Gastric protection:
- Proton pump inhibitor (omeprazole 20 mg daily or equivalent)
-
Cardiovascular risk management:
- Blood pressure control (target less than 130/80 mmHg)
- Statin therapy
- Management of hyperglycaemia (corticosteroid-induced diabetes common)
Alternative Induction Agents
Rituximab: [47]
- Anti-CD20 monoclonal antibody
- Indication: Cyclophosphamide contraindicated, intolerant, or refractory
- Evidence: Limited data in PAN specifically; extrapolated from ANCA vasculitis where non-inferior to cyclophosphamide (RAVE and RITUXVAS trials)
- Dosing:
- "Option 1: 375 mg/m² IV weekly × 4 doses"
- "Option 2: 1000 mg IV on days 0 and 14"
- Monitoring: B-cell depletion (CD19 count), hypogammaglobulinemia risk
- Infection risk: PJP prophylaxis recommended
- Some observational data suggests efficacy in refractory PAN [47]
Azathioprine: [6]
- Not recommended for induction (inferior to cyclophosphamide)
- Reserved for maintenance therapy
Methotrexate:
- Limited evidence in PAN
- Possible alternative for mild disease or maintenance
Mycophenolate mofetil:
- Limited data in PAN
- Some use as maintenance agent
Maintenance Therapy
After achieving remission (typically 3-6 months of induction), transition to maintenance therapy to prevent relapse:
Azathioprine (preferred): [6,48]
- Dose: 2 mg/kg/day orally (maximum 200 mg)
- Duration: 18-24 months minimum, often continued 36-48 months
- Monitor: FBC, liver function every 4-8 weeks
- Check TPMT genotype before starting (dose reduction or alternative if deficiency)
- Evidence: Standard of care based on ANCA vasculitis trials (CYCAZAREM)
Methotrexate (alternative): [49]
- Dose: 20-25 mg weekly orally or subcutaneously
- Contraindications: GFR less than 30 mL/min
- Monitoring: FBC, liver function, creatinine every 4-8 weeks
- Folic acid supplementation: 5 mg weekly (on different day from methotrexate)
Rituximab (emerging alternative):
- Repeat dosing every 6 months or based on B-cell reconstitution
- Emerging data in ANCA vasculitis (MAINRITSAN trials) [50]
- Limited specific data in PAN
Corticosteroid continuation:
- Continue low-dose prednisolone (5-10 mg daily) during maintenance
- Very slow taper over 18-24 months
- Attempt complete withdrawal if sustained remission ≥24 months
Duration of maintenance:
- Minimum 18-24 months
- Many experts continue 36-48 months
- Longer duration in relapsing disease or multiple poor prognostic factors
HBV-Associated PAN
HBV-associated PAN requires fundamentally different management focusing on viral suppression rather than prolonged immunosuppression. [5,51]
Principles
- Viral suppression is the primary goal
- Limited immunosuppression to control life-threatening manifestations
- Plasma exchange to remove circulating immune complexes
- Avoid prolonged immunosuppression which perpetuates HBV replication
Treatment Protocol [5,51]
Phase 1: Acute disease control (2-4 weeks)
Corticosteroids:
- Prednisolone 1 mg/kg/day or methylprednisolone 500-1000 mg IV × 3 days
- Short duration only (2-4 weeks maximum)
- Rapid taper once disease controlled
- Prolonged corticosteroids enhance viral replication
Plasma exchange:
- Removes circulating HBsAg-antibody immune complexes
- Protocol: 3-5 sessions over 7-10 days
- Exchange volume: 1-1.5 plasma volumes per session
- Replacement with fresh frozen plasma or albumin [52]
Cyclophosphamide (only if life-threatening, organ-threatening manifestations):
- Very limited use (1-3 pulses maximum)
- Consider only for severe manifestations unresponsive to above
Phase 2: Antiviral therapy (long-term)
Nucleos(t)ide analogues (first-line):
-
Entecavir: 0.5 mg daily orally (preferred)
- High barrier to resistance
- Excellent viral suppression
- Generally well-tolerated [53]
-
Tenofovir disoproxil fumarate (TDF): 300 mg daily orally
- High barrier to resistance
- Excellent viral suppression
- Monitor renal function (can cause renal toxicity) [53]
-
Tenofovir alafenamide (TAF): 25 mg daily orally
- Similar efficacy to TDF with improved renal and bone safety profile
- Preferred in patients with renal impairment or bone disease
Lamivudine (older agent, not recommended due to high resistance rates)
Alternative: Interferon-alpha (rarely used now):
- Pegylated interferon-alpha: 180 μg subcutaneously weekly
- Higher rates of seroconversion (HBsAg to anti-HBs)
- Poorly tolerated side effects
- Contraindicated in severe disease
- Rarely used since availability of oral antivirals [51]
Duration of antiviral therapy:
- Continue until HBsAg seroconversion (HBsAg negative, anti-HBs positive) PLUS 12 months
- If seroconversion not achieved, consider lifelong treatment
- Most patients require several years of treatment
- Monitor HBV DNA (aim undetectable), HBsAg, anti-HBs every 3-6 months
Outcomes
- Viral suppression achieved in > 90% with modern antivirals [5]
- Vasculitis remission rates 70-85% [51]
- Relapse risk low if viral suppression maintained
- Prognosis similar to non-HBV PAN if treated appropriately
Refractory Disease
Defined as failure to achieve remission after 3-6 months of standard therapy or progressive disease despite treatment. [6]
Management options:
- Verify diagnosis: Reconsider differential (infection, malignancy, other vasculitis)
- Assess adherence and drug levels if applicable
- Escalate/switch immunosuppression:
- Switch cyclophosphamide to rituximab (or vice versa)
- Increase corticosteroid dose
- Consider combination therapy (e.g., rituximab + cyclophosphamide)
- Plasma exchange: May provide benefit in severe refractory cases [54]
- Experimental agents:
- Tocilizumab (IL-6 inhibitor): Case reports [55]
- TNF inhibitors: Very limited data, mixed results
- IVIG: Case reports of benefit
Relapsing Disease
Relapse occurs in 10-30% of PAN patients during follow-up. [7]
Definition: Reappearance of disease activity after achieving remission.
Risk factors:
- Early treatment discontinuation
- Inadequate initial therapy
- HBV virological relapse (in HBV-associated PAN)
Management:
- Re-induction therapy: Restart corticosteroids ± cyclophosphamide/rituximab
- Assess for triggers: Infection, treatment non-adherence, HBV reactivation
- Extended maintenance: Consider longer duration or alternative maintenance agents
- Rituximab: May reduce relapse frequency (emerging data)
Specific Organ-Threatening Complications
Mesenteric ischaemia/infarction: [24]
- Urgent surgical consultation
- CT angiography to define extent
- High-dose IV corticosteroids (methylprednisolone 1000 mg daily × 3)
- Cyclophosphamide induction
- Surgical resection if perforation/infarction
- Consider plasma exchange
- Mortality risk high (30-50%) despite treatment
Cardiac involvement (MI, heart failure): [25]
- Standard acute coronary syndrome management
- High-dose corticosteroids + cyclophosphamide
- Coronary angiography (may show aneurysms; angioplasty challenging)
- Heart failure management as per guidelines
CNS vasculitis (stroke): [4]
- Neuroimaging (CT/MRI) to exclude haemorrhage
- High-dose IV corticosteroids
- Cyclophosphamide induction
- Standard stroke supportive care
Renal infarction: [23]
- Aggressive blood pressure control
- High-dose corticosteroids + cyclophosphamide
- Manage acute kidney injury (may require temporary dialysis)
Surgical Interventions
Indications:
- Bowel perforation or infarction: Resection of affected bowel
- Testicular infarction: Orchidectomy (also provides tissue for diagnosis)
- Severe digital ischaemia/gangrene: Amputation if non-viable
- Aneurysm rupture: Vascular surgical intervention
Considerations:
- Surgery in active vasculitis carries high morbidity
- Medical optimization first when possible
- Multidisciplinary approach essential
Monitoring During Treatment
Clinical assessment:
- Disease activity (BVAS) every visit
- Symptoms: constitutional, organ-specific
- Adverse effects of medications
Laboratory monitoring:
| Test | Frequency | Purpose |
|---|---|---|
| FBC | Every 2 weeks during CYC; every 4-8 weeks during maintenance | Leukopenia, anaemia monitoring |
| CRP/ESR | Every 2-4 weeks initially; every 4-8 weeks when stable | Disease activity marker |
| Creatinine/eGFR | Every 4 weeks initially; every 8-12 weeks when stable | Renal function |
| Urinalysis | Every 4-8 weeks | Haematuria (disease or CYC toxicity) |
| Liver function | Every 4-8 weeks | Hepatotoxicity monitoring |
| HBV DNA (if HBV+) | Every 3-6 months | Viral suppression |
| Glucose | Every 4-8 weeks | Steroid-induced diabetes |
Imaging:
- Repeat angiography: Not routinely required; reserve for clinical deterioration or diagnostic uncertainty
- Chest X-ray: Periodically to screen for opportunistic infections
9. Complications
Complications of PAN arise from both the disease itself and its treatment.
Disease-Related Complications
Organ infarction (20-40%): [4]
- Renal infarction: Acute kidney injury, chronic kidney disease
- Bowel infarction: Peritonitis, sepsis, death
- Myocardial infarction: Heart failure, arrhythmia, death
- Splenic infarction: Abdominal pain, splenic rupture (rare)
- Testicular infarction: Orchidectomy requirement
- Digital gangrene: Amputation requirement
Aneurysm rupture (5-10%): [4]
- Intra-abdominal bleeding from mesenteric/hepatic aneurysm rupture
- Presents with acute abdominal pain, hypotension, peritonism
- Requires emergency surgery
- High mortality (> 50%)
Bowel perforation (5-10%): [24]
- From transmural infarction
- Peritonitis and septic shock
- Requires emergency surgery
- Mortality 30-50%
Chronic kidney disease (30-40%): [23]
- Progressive renal impairment from cumulative ischaemic injury
- May progress to end-stage renal disease requiring dialysis/transplant
- Occurs in 10-20% of patients with renal involvement
Peripheral neuropathy sequelae (variable): [22]
- Permanent sensory deficits
- Persistent weakness (foot drop, wrist drop)
- Neuropathic pain syndrome
- Disability and reduced quality of life
Stroke (5-10%): [4]
- Ischaemic or haemorrhagic
- Permanent neurological deficits
- Associated with poor prognosis
Hypertensive complications: [23]
- Hypertensive emergency (encephalopathy, retinopathy)
- Left ventricular hypertrophy
- Increased cardiovascular risk
Treatment-Related Complications
Corticosteroid complications (very common): [46]
| Complication | Frequency | Prevention/Management |
|---|---|---|
| Infections | 20-30% | PJP prophylaxis; prompt treatment |
| Diabetes mellitus | 15-25% | Diet; metformin; insulin if required |
| Osteoporosis/fractures | 10-20% | Calcium/vitamin D; bisphosphonates; DEXA monitoring |
| Hypertension | 20-30% | Antihypertensive therapy |
| Weight gain | 50-70% | Dietary counselling |
| Mood changes | 10-30% | Psychological support; psychiatry if severe |
| Cataracts | 10-15% | Ophthalmology monitoring |
| Glaucoma | 5-10% | Ophthalmology monitoring |
| Avascular necrosis | 5-10% | Prompt imaging if joint pain; orthopaedic referral |
| Adrenal suppression | Universal | Gradual taper; stress-dose steroids for illness/surgery |
Cyclophosphamide complications [45]:
| Complication | Frequency | Prevention/Management |
|---|---|---|
| Opportunistic infections | 15-25% | PJP prophylaxis; prompt treatment |
| Haemorrhagic cystitis | 5-15% (higher with oral) | Hydration; urinalysis monitoring; cessation if occurs |
| Bladder cancer | 2-5% (cumulative dose-dependent) | Limit cumulative dose less than 36 g; urinalysis monitoring; cystoscopy if haematuria |
| Myelodysplasia/leukemia | 1-2% | Limit cumulative dose; FBC monitoring long-term |
| Infertility (women) | 50-90% (age-dependent) | GnRH agonists; fertility counselling; consider rituximab alternative |
| Infertility (men) | 50-90% | Sperm banking before treatment |
| Nausea/vomiting | 30-50% | Antiemetics (ondansetron) |
| Alopecia | 30-60% | Reversible; counselling |
Rituximab complications [47]:
| Complication | Frequency | Prevention/Management |
|---|---|---|
| Infusion reactions | 10-30% (first infusion) | Premedication (antihistamine, paracetamol); slower infusion |
| Infections | 15-20% | PJP prophylaxis; prompt treatment |
| Hypogammaglobulinemia | 5-10% | Monitor immunoglobulins; IVIG if recurrent infections |
| Progressive multifocal leukoencephalopathy (PML) | less than 0.1% | Vigilance for neurological symptoms; MRI if suspected |
| Hepatitis B reactivation | Variable | Screen HBsAg/anti-HBc; prophylactic antivirals if positive |
Azathioprine complications [48]:
- Bone marrow suppression: 5-10% (check TPMT; monitor FBC)
- Hepatotoxicity: 5-10% (monitor LFTs)
- Pancreatitis: 1-2% (check lipase if abdominal pain)
- Increased infection risk: 10-15%
- Increased skin cancer risk: Long-term use (sun protection)
10. Prognosis
Survival Outcomes
The prognosis of PAN has improved dramatically with modern immunosuppressive therapy. [7]
Historical outcomes (pre-corticosteroid era):
- 5-year survival: ~10-15%
- Median survival: less than 1 year
- Death from untreated vasculitis complications
Modern outcomes (with immunosuppression): [7]
- 5-year survival: 75-85%
- 10-year survival: 60-70%
- Mortality predominantly from disease complications in first 2 years; later mortality from treatment complications and cardiovascular disease
Prognostic Factors
Poor prognostic factors (Five Factor Score components): [41]
- Age > 65 years
- Cardiac involvement
- Gastrointestinal involvement (especially infarction/perforation)
- Renal insufficiency (creatinine > 150 μmol/L)
- FFS ≥2: 5-year mortality 40-50% without intensive treatment
Favourable prognostic factors:
- Young age
- Limited disease (FFS = 0)
- Early diagnosis and treatment
- HBV-associated PAN with viral suppression
- Absence of cardiac or GI involvement
Causes of Death
Early mortality (first 2 years): [7]
- Active vasculitis complications (50-60% of deaths):
- Mesenteric infarction/perforation
- Myocardial infarction
- Stroke
- Renal failure
- Multiorgan failure
- Infections (30-40% of deaths):
- Opportunistic infections (PJP, CMV, fungi)
- Bacterial sepsis
- Related to intensive immunosuppression
Late mortality (> 2 years):
- Cardiovascular disease (30-40%)
- Infections (20-30%)
- Malignancy (10-15%, including treatment-related)
- Chronic kidney disease complications (10%)
Relapse Rates
- Overall relapse rate: 10-30% [7]
- Most relapses occur within first 2 years after treatment
- Relapse risk reduced with extended maintenance therapy
- HBV-associated PAN: Lower relapse if viral suppression maintained
Long-Term Morbidity
Chronic kidney disease: [23]
- Develops in 30-40% of patients with renal involvement
- 10-20% progress to end-stage renal disease
- Requires long-term nephrology follow-up
Peripheral neuropathy: [22]
- Incomplete recovery common
- Persistent sensory deficits, weakness, neuropathic pain
- Rehabilitation and supportive care important
Cardiovascular morbidity:
- Increased cardiovascular risk from disease, hypertension, corticosteroids
- Long-term cardiovascular risk reduction strategies essential
Treatment-related morbidity:
- Cumulative corticosteroid effects (osteoporosis, diabetes, weight gain)
- Cyclophosphamide-related bladder cancer, myelodysplasia (rare)
- Increased infection susceptibility
Quality of Life
Studies demonstrate reduced health-related quality of life in PAN patients compared to general population: [20]
- Fatigue: Persistent in 40-60%
- Pain: Chronic pain from neuropathy, musculoskeletal involvement
- Physical function: Reduced in those with residual neuropathy or organ damage
- Psychological impact: Depression, anxiety related to chronic disease and treatment
11. Prevention and Screening
Primary Prevention
Hepatitis B vaccination: [5]
- Universal HBV vaccination has dramatically reduced HBV-associated PAN incidence
- Recommended for all infants and at-risk adults
- Most important primary prevention strategy
Blood product screening:
- Universal screening of blood products for HBV has reduced transmission-associated PAN
No other primary prevention strategies identified for idiopathic PAN given unclear aetiology.
Secondary Prevention (Relapse Prevention)
Adequate duration of maintenance therapy: [6]
- Minimum 18-24 months, preferably 36-48 months
- Reduces relapse risk from ~40% to 10-20%
HBV viral suppression: [51]
- Maintain undetectable HBV DNA with antivirals
- Prevents virological and vasculitis relapse
Regular monitoring:
- Clinical assessment every 3-6 months during remission
- CRP/ESR every 3-6 months
- Creatinine, urinalysis every 3-6 months
- Early detection of relapse allows prompt intervention
Cardiovascular risk reduction:
- Blood pressure control
- Lipid management (statins)
- Diabetes control
- Smoking cessation
- Regular cardiovascular screening
Tertiary Prevention (Complication Prevention)
Infection prevention:
- PJP prophylaxis during intensive immunosuppression [44]
- Vaccination (pneumococcal, influenza—avoid live vaccines during immunosuppression)
- Prompt treatment of infections
Osteoporosis prevention: [46]
- Calcium and vitamin D supplementation
- Bisphosphonates for patients on long-term corticosteroids
- DEXA scanning at baseline and periodically
Gastric protection:
- Proton pump inhibitors during high-dose corticosteroids
Fertility preservation: [45]
- Sperm banking for men before cyclophosphamide
- GnRH agonists for women during cyclophosphamide
- Consider rituximab as alternative if fertility priority
Bladder cancer prevention:
- Limit cyclophosphamide cumulative dose to less than 36 g
- Urinalysis monitoring during and after treatment
- Prompt investigation of haematuria
Screening Recommendations
For patients with PAN:
- Annual cardiovascular risk assessment
- Annual urinalysis and renal function (lifelong)
- Blood pressure monitoring (every visit)
- Glucose monitoring (at least annually while on corticosteroids)
- DEXA scan every 1-2 years while on corticosteroids
- Malignancy screening as per age-appropriate guidelines (higher vigilance given immunosuppression)
For HBV-associated PAN:
- HBV DNA monitoring every 3-6 months while on antivirals
- Liver ultrasound every 6 months (hepatocellular carcinoma screening)
- Alpha-fetoprotein every 6 months
12. Key Guidelines and Consensus Statements
Chapel Hill Consensus Conference Nomenclature (2012): [2]
- International consensus on vasculitis classification
- Defines PAN as necrotizing medium/small artery vasculitis without glomerulonephritis, not ANCA-associated
- Essential for correct diagnosis and classification
American College of Rheumatology Classification Criteria (1990): [40]
- Classification criteria requiring ≥3 of 10 criteria
- Sensitivity 82%, specificity 87%
- Used for research classification; clinical diagnosis requires integrated assessment
EULAR Recommendations for Vasculitis Management: [6]
- Evidence-based recommendations for induction and maintenance therapy
- Guides treatment approach based on disease severity
- Recommends corticosteroids alone for mild disease, combination therapy for severe disease
French Vasculitis Study Group Protocols: [51]
- Specific protocols for HBV-associated PAN management
- Emphasizes antiviral therapy, plasma exchange, limited immunosuppression
British Society for Rheumatology Guidelines on ANCA-Associated Vasculitis: [56]
- While focused on ANCA vasculitis, many principles applicable to PAN
- Guidance on cyclophosphamide use, infection prophylaxis, monitoring
13. Examination Focus
Common Exam Questions
-
"How would you distinguish PAN from microscopic polyangiitis?"
- ANCA negative vs positive
- Medium arteries vs small vessels (capillaries)
- Absence vs presence of glomerulonephritis
- Aneurysms on angiography vs normal angiography
- Pulmonary sparing vs pulmonary involvement
-
"What is the significance of ANCA testing in suspected PAN?"
- Must be negative for PAN diagnosis (Chapel Hill criteria)
- ANCA positivity indicates ANCA-associated vasculitis (MPA, GPA, EGPA)
- Both c-ANCA (PR3) and p-ANCA (MPO) should be negative
- Mandatory test in diagnostic workup
-
"Describe the management of HBV-associated PAN."
- Differs fundamentally from non-HBV PAN
- Short-course corticosteroids (2-4 weeks) for acute control
- Plasma exchange to remove immune complexes
- Antiviral therapy (entecavir or tenofovir) as primary treatment
- Avoid prolonged immunosuppression (promotes viral replication)
- Goal: viral suppression and seroconversion
-
"What are the characteristic angiographic findings in PAN?"
- Microaneurysms (1-5 mm) at arterial branch points
- "Beading" or "rosary" appearance
- Arterial stenoses and irregularity
- Distribution: renal, hepatic, mesenteric arteries most commonly
- Highly specific when present in appropriate clinical context
-
"What is the Five Factor Score and how does it guide treatment?"
- Prognostic scoring system for PAN
- Factors: Age > 65, cardiac symptoms, GI involvement, renal insufficiency
- FFS = 0: Mild disease, consider corticosteroid monotherapy
- FFS ≥1: Moderate-severe disease, requires combination therapy (corticosteroids + cyclophosphamide)
- Predicts mortality and guides treatment intensity
Viva Points
Opening statement for PAN viva:
"Polyarteritis nodosa is a rare necrotizing vasculitis affecting medium-sized muscular arteries, classified under the Chapel Hill Consensus as ANCA-negative vasculitis without glomerulonephritis. The annual incidence is approximately 2-9 per million, with peak onset in the fifth to sixth decade. It presents with constitutional symptoms and multisystem involvement including peripheral neuropathy—classically mononeuritis multiplex—renal disease with hypertension and infarction but not glomerulonephritis, gastrointestinal involvement with mesenteric ischaemia, and cutaneous manifestations such as livedo reticularis and nodules. Approximately 5-10% of cases are associated with hepatitis B infection, though this has declined with vaccination."
Structured approach to PAN diagnosis:
"I would approach suspected PAN systematically. First, clinical assessment for characteristic features: constitutional symptoms, mononeuritis multiplex, livedo reticularis, hypertension, abdominal pain, and testicular pain. Second, essential laboratory tests: ANCA must be negative, HBV serology mandatory, elevated inflammatory markers expected, and urinalysis should show bland sediment without glomerulonephritis. Third, imaging with CT or MR angiography of renal and mesenteric vessels looking for characteristic microaneurysms and stenoses. Fourth, consider tissue biopsy—combined sural nerve and muscle biopsy has highest yield at 70-80%—showing necrotizing vasculitis of medium arteries. The diagnosis requires integration of clinical features, ANCA negativity, characteristic imaging or histology, and exclusion of mimics."
Treatment approach:
"Management depends on disease severity using the Five Factor Score and HBV status. For non-HBV PAN with FFS of zero—mild disease without organ-threatening features—I would use corticosteroid monotherapy with prednisolone 1 mg/kg daily, tapered over 12-18 months. For FFS of one or more—indicating moderate to severe disease—combination therapy with corticosteroids plus cyclophosphamide is required, typically IV pulse cyclophosphamide 15 mg/kg every 2-3 weeks for 3-6 months, then transitioning to maintenance with azathioprine 2 mg/kg daily for 18-24 months. I would provide PJP prophylaxis with co-trimoxazole and bone protection with calcium, vitamin D, and bisphosphonates. For HBV-associated PAN, the approach differs: short-course corticosteroids for acute control, plasma exchange to remove immune complexes, and antiviral therapy with entecavir or tenofovir as primary treatment, avoiding prolonged immunosuppression which would enhance viral replication."
Key distinguishing features from ANCA vasculitis:
"PAN differs from ANCA-associated vasculitides in several critical ways. First, ANCA must be negative in PAN by definition. Second, vessel size: PAN affects medium muscular arteries whereas microscopic polyangiitis affects capillaries and small vessels. Third, glomerulonephritis is absent in PAN but prominent in MPA and GPA. Fourth, pulmonary involvement is absent in classic PAN—pulmonary involvement suggests ANCA vasculitis. Fifth, angiography shows characteristic microaneurysms in PAN but is typically normal in ANCA vasculitis. Finally, PAN may be associated with HBV infection whereas ANCA vasculitides are not."
Red flag complications requiring urgent intervention:
"Life-threatening complications of PAN include mesenteric ischaemia or infarction presenting with severe abdominal pain—requires urgent CT angiography, high-dose IV corticosteroids, cyclophosphamide, and surgical consultation for possible bowel resection. Myocardial infarction from coronary arteritis requires standard ACS management plus immunosuppression. Acute stroke from CNS vasculitis requires neuroimaging, high-dose corticosteroids, and cyclophosphamide. Aneurysm rupture with intra-abdominal bleeding requires emergency vascular surgery. These complications carry mortality rates of 30-50% despite treatment and represent medical emergencies requiring multidisciplinary care in high-dependency or intensive care settings."
Common Mistakes in Exams
❌ Mistake 1: Stating that PAN can be ANCA-positive
- Correction: ANCA must be negative by Chapel Hill definition; ANCA positivity indicates AAV not PAN
❌ Mistake 2: Describing glomerulonephritis as a feature of PAN
- Correction: Glomerulonephritis is characteristically absent; its presence suggests MPA
❌ Mistake 3: Missing the association with hepatitis B
- Correction: Always check HBV serology; 5-10% are HBV-associated requiring different treatment
❌ Mistake 4: Using prolonged immunosuppression for HBV-associated PAN
- Correction: HBV-PAN requires antivirals as primary treatment with short-course steroids and plasma exchange
❌ Mistake 5: Not mentioning mononeuritis multiplex
- Correction: Mononeuritis multiplex is one of the most characteristic features (50-70%)
❌ Mistake 6: Suggesting lung involvement
- Correction: Pulmonary involvement is absent in classic PAN; presence suggests AAV
❌ Mistake 7: Recommending corticosteroid monotherapy for severe disease (FFS ≥1)
- Correction: Severe disease requires combination therapy with cyclophosphamide or rituximab
❌ Mistake 8: Forgetting PJP prophylaxis
- Correction: Essential during cyclophosphamide therapy; co-trimoxazole is standard
Model Answer for Case Scenario
Scenario: "A 52-year-old man presents with 3-month history of weight loss, fever, and progressive painful peripheral neuropathy affecting right foot and left hand. Examination shows foot drop, wrist drop, and livedo reticularis on legs. BP 165/95. Blood: Hb 105, WBC 12.5, CRP 98. Creatinine 145. Urinalysis: protein +, blood +, no casts. How would you investigate and manage?"
Model answer:
"This presentation is highly suggestive of polyarteritis nodosa with characteristic mononeuritis multiplex, constitutional symptoms, hypertension, and livedo reticularis.
My investigations would include:
First, essential blood tests: ANCA testing—must be negative for PAN diagnosis; hepatitis B serology including HBsAg and anti-HBc as 5-10% of PAN is HBV-associated; ESR which I expect to be markedly elevated; complement levels which should be normal; and hepatitis C and HIV serology to exclude other causes.
Second, imaging: CT or MR angiography of renal and mesenteric vessels looking for characteristic microaneurysms and stenoses at arterial branch points, which would show a beading or rosary appearance and be highly specific for PAN.
Third, neurophysiology: EMG and nerve conduction studies to characterize the neuropathy, which I expect would show asymmetric multifocal axonal neuropathy consistent with mononeuritis multiplex.
Fourth, consider tissue biopsy: Given the peripheral neuropathy, combined sural nerve and gastrocnemius muscle biopsy would have diagnostic yield of 70-80%, showing necrotizing vasculitis of medium arteries with fibrinoid necrosis.
Regarding management, this patient has several poor prognostic features including renal impairment giving a Five Factor Score of at least 1, indicating moderate-severe disease requiring combination immunosuppression. I would commence:
Induction therapy with prednisolone 1 mg/kg daily—approximately 70-80 mg for this patient—plus IV pulse cyclophosphamide 15 mg/kg every 2-3 weeks, dose-adjusted for renal function. Duration of induction would be 3-6 months with aim to achieve remission.
Supportive measures including co-trimoxazole for PJP prophylaxis, calcium and vitamin D with bisphosphonate for bone protection, proton pump inhibitor for gastric protection, and aggressive blood pressure control targeting below 130/80.
After achieving remission at 3-6 months, I would transition to maintenance therapy with azathioprine 2 mg/kg daily—checking TPMT first—and continue for minimum 18-24 months while slowly tapering corticosteroids.
I would monitor treatment response with clinical assessment, serial CRP/ESR, renal function, and urinalysis every 2-4 weeks initially. With this approach, I would expect 80-90% remission rate and 5-year survival exceeding 80%. The main risks are treatment complications including infections and corticosteroid adverse effects, requiring vigilant monitoring."
References
-
Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11. doi:10.1002/art.37715
-
Pagnoux C, Seror R, Henegar C, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis Rheum. 2010;62(2):616-626. doi:10.1002/art.27240
-
Hernández-Rodríguez J, Alba MA, Prieto-González S, Cid MC. Diagnosis and classification of polyarteritis nodosa. J Autoimmun. 2014;48-49:84-89. doi:10.1016/j.jaut.2014.01.029
-
De Virgilio A, Greco A, Magliulo G, et al. Polyarteritis nodosa: A contemporary overview. Autoimmun Rev. 2016;15(6):564-570. doi:10.1016/j.autrev.2016.02.015
-
Guillevin L, Mahr A, Callard P, et al. Hepatitis B virus-associated polyarteritis nodosa: clinical characteristics, outcome, and impact of treatment in 115 patients. Medicine (Baltimore). 2005;84(5):313-322. doi:10.1097/01.md.0000180792.80212.5e
-
Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68(3):310-317. doi:10.1136/ard.2008.088096
-
Bourgarit A, Le Toumelin P, Pagnoux C, et al. Deaths occurring during the first year after treatment onset for polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: a retrospective analysis of causes and factors predictive of mortality based on 595 patients. Medicine (Baltimore). 2005;84(5):323-330. doi:10.1097/01.md.0000180793.80078.ca
-
Mohammad AJ, Jacobsson LT, Mahr AD, Sturfelt G, Segelmark M. Prevalence of Wegener's granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford). 2007;46(8):1329-1337. doi:10.1093/rheumatology/kem107
-
Mahr A, Guillevin L, Poissonnet M, Aymé S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum. 2004;51(1):92-99. doi:10.1002/art.20077
-
Mahr A, Katsahian S, Varet H, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. 2013;72(6):1003-1010. doi:10.1136/annrheumdis-2012-201750
-
Trepo C, Guillevin L. Polyarteritis nodosa and extrahepatic manifestations of HBV infection: the case against autoimmune intervention in pathogenesis. J Autoimmun. 2001;16(3):269-274. doi:10.1006/jaut.2000.0502
-
Ozen S, Anton J, Arisoy N, et al. Juvenile polyarteritis: results of a multicenter survey of 110 children. J Pediatr. 2004;145(4):517-522. doi:10.1016/j.jpeds.2004.06.046
-
Guillevin L, Lhote F, Cohen P, et al. Polyarteritis nodosa related to hepatitis B virus. A prospective study with long-term observation of 41 patients. Medicine (Baltimore). 1995;74(5):238-253. doi:10.1097/00005792-199509000-00002
-
Choi HK, Merkel PA, Walker AM, Niles JL. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43(2):405-413. doi:10.1002/1529-0131(200002)43:2less than 405::AID-ANR23> 3.0.CO;2-5
-
Hasler P, Kistler H, Gerber H. Vasculitides in hairy cell leukemia. Semin Arthritis Rheum. 1995;25(2):134-142. doi:10.1016/s0049-0172(95)80027-2
-
Ozdogan H, Arisoy N, Kasapçapur O, et al. Vasculitis in familial Mediterranean fever. J Rheumatol. 1997;24(2):323-327.
-
Kawakami T, Yamazaki M, Mizoguchi M, Soma Y. High titer of serum antiphospholipid antibody levels in adult Henoch-Schönlein purpura and cutaneous leukocytoclastic angiitis. Arthritis Rheum. 2008;59(4):561-567. doi:10.1002/art.23439
-
Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9(12):731-740. doi:10.1038/nrrheum.2013.161
-
Cid MC, Hernández-Rodríguez J, Robert J, et al. Tissue and serum matrix metalloproteinase-9 in giant cell arteritis: correlation with systemic cytokine profile. Rheumatology (Oxford). 2006;45(2):171-175. doi:10.1093/rheumatology/kei128
-
Basu N, McClean A, Harper L, et al. Explaining fatigue in ANCA-associated vasculitis. Rheumatology (Oxford). 2013;52(9):1680-1685. doi:10.1093/rheumatology/ket191
-
Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol. 1997;136(5):706-713.
-
Collins MP, Periquet MI. Isolated vasculitis of the peripheral nervous system. Clin Exp Rheumatol. 2008;26(3 Suppl 49):S118-S130.
-
Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512-1523. doi:10.1056/NEJM199711203372106
-
Ha HK, Lee SH, Rha SE, et al. Radiologic features of vasculitis involving the gastrointestinal tract. Radiographics. 2000;20(3):779-794. doi:10.1148/radiographics.20.3.g00ma02779
-
Morgan JM, Raposo L, Gibson DG. Cardiac involvement in polyarteritis nodosa. Br Heart J. 1989;62(6):459-464. doi:10.1136/hrt.62.6.459
-
Warshauer DM, Lee JK. Imaging manifestations of abdominal sarcoidosis. AJR Am J Roentgenol. 2004;182(1):15-28. doi:10.2214/ajr.182.1.1820015
-
Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology and prognosis. Q J Med. 1985;56(220):467-483.
-
Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116(6):488-498. doi:10.7326/0003-4819-116-6-488
-
Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990;33(8):1094-1100. doi:10.1002/art.1780330806
-
McCrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation. 2017;135(17):e927-e999. doi:10.1161/CIR.0000000000000484
-
Cacoub P, Comarmond C, Domont F, Savey L, Saadoun D. Cryoglobulinemia Vasculitis. Am J Med. 2015;128(9):950-955. doi:10.1016/j.amjmed.2015.02.017
-
Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein Purpura in adults: outcome and prognostic factors. J Am Soc Nephrol. 2002;13(5):1271-1278. doi:10.1097/01.asn.0000013883.99976.22
-
Criteria for diagnosis of Behçet's disease. International Study Group for Behçet's Disease. Lancet. 1990;335(8697):1078-1080.
-
Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med. 1994;120(11):919-929. doi:10.7326/0003-4819-120-11-199406010-00004
-
Murdoch DR, Corey GR, Hoen B, et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med. 2009;169(5):463-473. doi:10.1001/archinternmed.2008.603
-
Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116(3):298-304. doi:10.1161/CIRCULATIONAHA.106.680991
-
Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295-306. doi:10.1111/j.1538-7836.2006.01753.x
-
Olin JW, Gornik HL, Bacharach JM, et al. Fibromuscular dysplasia: state of the science and critical unanswered questions: a scientific statement from the American Heart Association. Circulation. 2014;129(9):1048-1078. doi:10.1161/01.cir.0000442577.96802.8c
-
Ebert EC, Hagspiel KD, Nagar M, Schlesinger N. Gastrointestinal involvement in polyarteritis nodosa. Clin Gastroenterol Hepatol. 2008;6(9):960-966. doi:10.1016/j.cgh.2008.04.004
-
Lightfoot RW Jr, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum. 1990;33(8):1088-1093. doi:10.1002/art.1780330805
-
Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P. The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore). 2011;90(1):19-27. doi:10.1097/MD.0b013e318205a4c6
-
Mukhtyar C, Lee R, Brown D, et al. Modification and validation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis. 2009;68(12):1827-1832. doi:10.1136/ard.2008.101279
-
de Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150(10):670-680. doi:10.7326/0003-4819-150-10-200905190-00004
-
Stern A, Green H, Paul M, Vidal L, Leibovici L. Prophylaxis for Pneumocystis pneumonia (PCP) in non-HIV immunocompromised patients. Cochrane Database Syst Rev. 2014;(10):CD005590. doi:10.1002/14651858.CD005590.pub3
-
Clowse ME, Copland SC, Hsieh TC, et al. Ovarian reserve diminished by oral cyclophosphamide therapy for granulomatosis with polyangiitis (Wegener's). Arthritis Care Res (Hoboken). 2011;63(12):1777-1781. doi:10.1002/acr.20605
-
Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken). 2010;62(11):1515-1526. doi:10.1002/acr.20295
-
Cartin-Ceba R, Keogh KA, Specks U, Sethi S, Fervenza FC. Rituximab for the treatment of Churg-Strauss syndrome with renal involvement. Nephrol Dial Transplant. 2011;26(9):2865-2871. doi:10.1093/ndt/gfr143
-
Pagnoux C, Mahr A, Hamidou MA, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 2008;359(26):2790-2803. doi:10.1056/NEJMoa0802311
-
De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005;52(8):2461-2469. doi:10.1002/art.21142
-
Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371(19):1771-1780. doi:10.1056/NEJMoa1404231
-
Guillevin L, Lhote F, Cohen P, et al. Corticosteroids plus pulse cyclophosphamide and plasma exchanges versus corticosteroids plus pulse cyclophosphamide alone in the treatment of polyarteritis nodosa and Churg-Strauss syndrome patients with factors predicting poor prognosis. A prospective, randomized trial in sixty-two patients. Arthritis Rheum. 1995;38(11):1638-1645. doi:10.1002/art.1780381117
-
Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996;75(1):17-28. doi:10.1097/00005792-199601000-00003
-
Terrault NA, Lok ASF, McMahon BJ, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67(4):1560-1599. doi:10.1002/hep.29800
-
Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18(7):2180-2188. doi:10.1681/ASN.2007010090
-
Unizony S, Arias-Urdaneta L, Miloslavsky E, et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res (Hoboken). 2012;64(11):1720-1729. doi:10.1002/acr.21750
-
Ntatsaki E, Carruthers D, Chakravarty K, et al. BSR and BHPR guideline for the management of adults with ANCA-associated vasculitis. Rheumatology (Oxford). 2014;53(12):2306-2309. doi:10.1093/rheumatology/ket445
Last Updated: 2026-01-05
Frequently asked questions
Quick clarifications for common clinical and exam-facing questions.
When should I seek emergency care for polyarteritis nodosa?
Seek immediate emergency care if you experience any of the following warning signs: Mesenteric ischaemia and bowel perforation, Acute renal failure with renal infarction, Severe peripheral neuropathy with muscle weakness, Cardiac involvement with MI or heart failure, Testicular infarction, CNS involvement with stroke or seizures.
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Vasculitis Classification
- Immune Complex Disease
Differentials
Competing diagnoses and look-alikes to compare.
- Microscopic Polyangiitis
- Granulomatosis with Polyangiitis
- Eosinophilic Granulomatosis with Polyangiitis
- Kawasaki Disease