Phaeochromocytoma and Paraganglioma
Phaeochromocytoma is a rare catecholamine-secreting tumour arising from chromaffin cells of the adrenal medulla. When arising from extra-adrenal sympathetic ganglia (paraganglia), the tumour is termed paraganglioma....
What matters first
Phaeochromocytoma is a rare catecholamine-secreting tumour arising from chromaffin cells of the adrenal medulla. When arising from extra-adrenal sympathetic ganglia (paraganglia), the tumour is termed paraganglioma....
Hypertensive crisis or paroxysmal severe hypertension
7 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Hypertensive crisis or paroxysmal severe hypertension
- Classical triad: headache, sweating, palpitations
- Resistant hypertension (less than 3 antihypertensives)
- Hypertension in young patient (less than 40 years)
Linked comparisons
Differentials and adjacent topics worth opening next.
- Essential Hypertension
- Anxiety Disorders and Panic Attacks
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Phaeochromocytoma and Paraganglioma
1. Clinical Overview
Summary
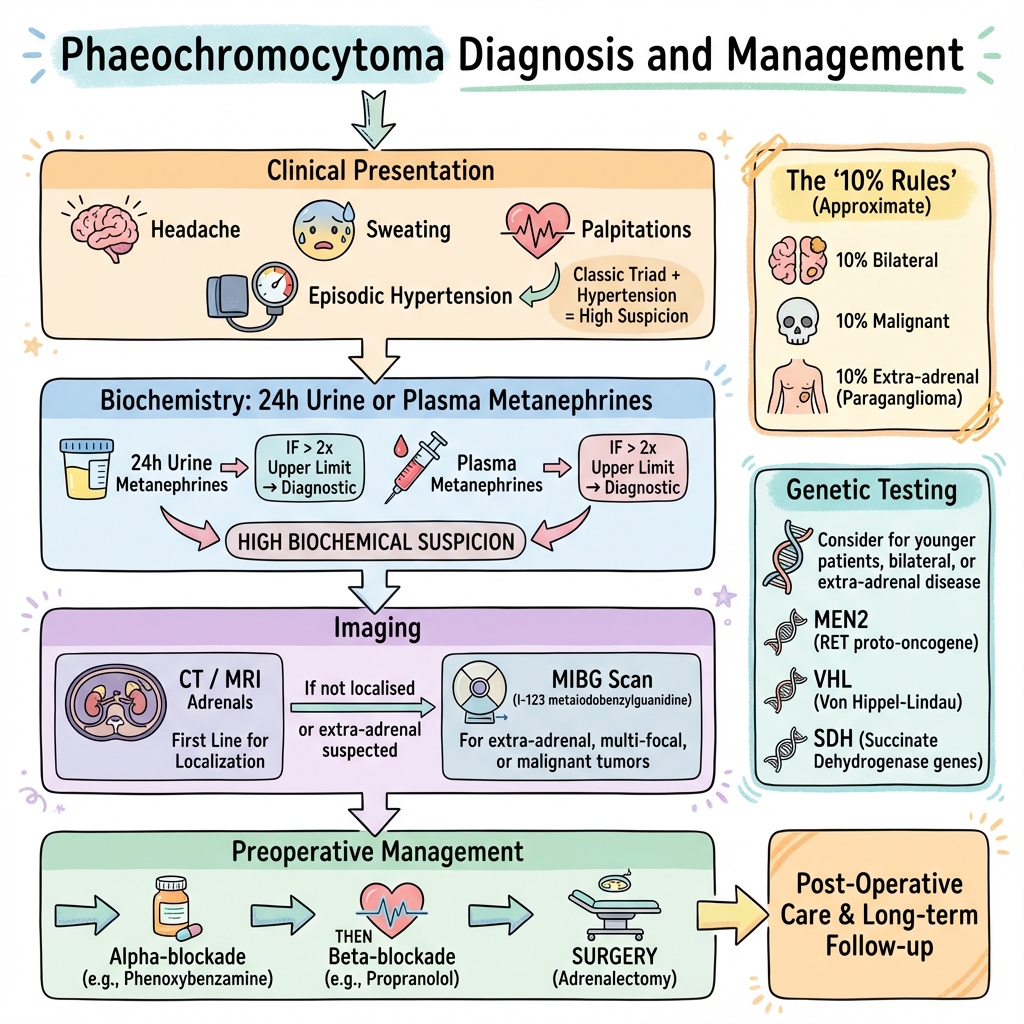
Phaeochromocytoma is a rare catecholamine-secreting tumour arising from chromaffin cells of the adrenal medulla. When arising from extra-adrenal sympathetic ganglia (paraganglia), the tumour is termed paraganglioma. These neuroendocrine tumours produce excess catecholamines (predominantly noradrenaline and adrenaline), causing paroxysmal or sustained hypertension, headaches, sweating, and palpitations—the classical triad of symptoms. [1,2]
Although historically described by the "rule of 10s" (10% bilateral, 10% malignant, 10% extra-adrenal, 10% familial), contemporary genetic studies have revealed that up to 40% of cases are associated with hereditary syndromes including Multiple Endocrine Neoplasia type 2 (MEN2), von Hippel-Lindau disease (VHL), and succinate dehydrogenase (SDH) mutations. [3,4] This high prevalence of germline mutations has fundamentally changed clinical practice, with genetic testing now recommended for all patients diagnosed with phaeochromocytoma or paraganglioma.
Diagnosis relies on biochemical confirmation through measurement of plasma or 24-hour urinary fractionated metanephrines, which demonstrate sensitivity exceeding 95% when levels are more than twice the upper limit of normal. [5,6] Following biochemical confirmation, anatomical localisation is achieved through CT or MRI imaging, with functional imaging (MIBG scintigraphy or 68Ga-DOTATATE PET/CT) reserved for specific scenarios. [7] Treatment requires meticulous preoperative preparation with alpha-adrenergic blockade followed by beta-blockade before definitive surgical resection. The critical management principle is that beta-blockade must NEVER precede alpha-blockade, as this can precipitate unopposed alpha-stimulation and hypertensive crisis. [1,8]
Key Facts
- Definition: Catecholamine-secreting tumour arising from chromaffin cells (adrenal medulla) or paraganglia (extra-adrenal)
- Incidence: 2-8 per million per year; accounts for 0.1-0.6% of hypertensive patients [9]
- Peak Demographics: 30-50 years; no significant sex predominance
- Classical Triad: Episodic headache (80%), profuse sweating (70%), and palpitations (70%) [2]
- Pathognomonic Finding: Plasma or urinary metanephrines > 2× upper limit of normal
- Gold Standard Investigation: Plasma free metanephrines (sensitivity 96-99%, specificity 89-95%) [5,6]
- Genetic Associations: 40% hereditary; test all patients for RET, VHL, SDHB/C/D, NF1 mutations [3,4]
- Malignancy Rate: 10-17% (higher in SDHB mutations and extra-adrenal tumours) [10]
- First-line Treatment: Preoperative alpha-blockade (phenoxybenzamine or doxazosin), then beta-blockade, then surgical resection [1,8]
- Prognosis: 90% cure with complete surgical resection; lifelong follow-up required for recurrence [11]
Clinical Pearls
Diagnostic Pearl: The "5 Ps" mnemonic - Pressure (hypertension), Pain (headache), Perspiration (sweating), Palpitations, Pallor (not flushing). The presence of the classical triad makes the diagnosis highly likely.
Treatment Pearl: ALWAYS alpha-block BEFORE beta-block. Initiating beta-blockade alone causes unopposed alpha-receptor stimulation, precipitating severe vasoconstriction and hypertensive crisis. This is a life-threatening error.
Emergency Pearl: Phaeochromocytoma crisis (severe hypertension with end-organ damage) is managed with intravenous phentolamine (alpha-blocker) or sodium nitroprusside, NOT beta-blockers alone.
Biochemical Pearl: Metanephrines are continuously produced within the tumour via catechol-O-methyltransferase (COMT), making them more sensitive markers than catecholamines, which are released episodically. Plasma metanephrines can be measured regardless of symptom timing.
Genetic Pearl: The hereditary syndromes follow different patterns - MEN2 (RET mutation) has bilateral phaeochromocytomas in 50%; SDHB mutations confer 30-70% malignancy risk; VHL typically presents earlier (20s-30s). Genetic testing is essential for all patients and enables cascade screening of relatives. [3,4]
Why This Matters Clinically
Phaeochromocytoma represents a curable cause of secondary hypertension but carries significant mortality if unrecognised. Undiagnosed phaeochromocytoma poses life-threatening risks during surgery, anaesthesia, pregnancy, or childbirth due to catecholamine surges causing hypertensive crisis, myocardial infarction, stroke, or arrhythmias. [12] The incidental discovery of adrenal masses on imaging ("adrenal incidentalomas") is increasingly common, occurring in 4-7% of abdominal CT scans, and biochemical screening for phaeochromocytoma is mandatory in all such cases. [13] Early diagnosis enables safe preoperative preparation and prevents catastrophic intraoperative complications.
2. Epidemiology
Incidence and Prevalence
Phaeochromocytoma is rare, with an estimated annual incidence of 2-8 cases per million population. [9] Autopsy studies suggest the condition is significantly underdiagnosed, with prevalence estimates of 0.05-0.1% in the general population. Among hypertensive patients, the prevalence is 0.1-0.6%, rising to 4-5% in patients with adrenal incidentalomas. [13,14]
| Epidemiological Feature | Value | Reference |
|---|---|---|
| Annual incidence | 2-8 per million | [9] |
| Prevalence in hypertensive patients | 0.1-0.6% | [14] |
| Prevalence in adrenal incidentalomas | 4-5% | [13] |
| Median age at diagnosis | 40-50 years | [2] |
| Male:female ratio | Approximately 1:1 | [2] |
| Hereditary cases | 30-40% | [3,4] |
Demographics
Phaeochromocytoma can occur at any age but peak incidence is in the 4th-5th decades. Hereditary syndromes often present earlier (20s-30s), particularly VHL disease. There is no significant sex predominance in sporadic cases, though certain hereditary syndromes show sex-specific patterns (e.g., MEN2A affects males and females equally).
The "Rule of 10s"
- Historical Context and Modern Updates
The traditional "rule of 10s" was a helpful mnemonic but modern data has revised several of these figures:
| Historical "Rule" | Traditional Teaching | Modern Evidence | Updated Estimate |
|---|---|---|---|
| 10% extra-adrenal (paraganglioma) | 10% | Confirmed | 10-15% [2] |
| 10% bilateral | 10% | Higher in hereditary syndromes | 10% sporadic; 50% in MEN2 [15] |
| 10% malignant | 10% | Variable by genotype | 10-17% overall; up to 70% in SDHB [10] |
| 10% familial | 10% | Significantly underestimated | 30-40% with germline mutations [3,4] |
| 10% children | 10% | Confirmed | 10% occur in paediatric population |
| 10% normotensive | Rare | Usually dopamine-secreting | less than 5% truly normotensive |
Hereditary Syndromes and Genetic Associations
Approximately 30-40% of phaeochromocytomas and paragangliomas are now recognised to be associated with germline mutations in over 20 susceptibility genes. [3,4] The major hereditary syndromes include:
| Syndrome | Gene | Inheritance | Key Features | Phaeochromocytoma Risk |
|---|---|---|---|---|
| MEN2A/2B | RET | Autosomal dominant | Medullary thyroid cancer (95%), primary hyperparathyroidism (20-30% in MEN2A) | 50% develop bilateral phaeochromocytomas [15] |
| Von Hippel-Lindau (VHL) | VHL | Autosomal dominant | Cerebellar/spinal haemangioblastomas, renal cell carcinoma, pancreatic neuroendocrine tumours | 10-20% develop phaeochromocytoma (higher in VHL type 2) [16] |
| Neurofibromatosis Type 1 (NF1) | NF1 | Autosomal dominant | Café-au-lait spots, neurofibromas, optic gliomas | 1-5% develop phaeochromocytoma [17] |
| SDH-associated syndromes | SDHB, SDHC, SDHD, SDHAF2 | Autosomal dominant | Paragangliomas (especially head/neck) | SDHB: 30-70% malignant; SDHD: rarely malignant [4,10] |
| Familial paraganglioma syndromes | Multiple SDH genes | Autosomal dominant | Predominantly extra-adrenal paragangliomas | Variable penetrance |
Clinical Implication: Genetic testing is now recommended for ALL patients diagnosed with phaeochromocytoma or paraganglioma, regardless of family history or age, due to the high prevalence of germline mutations. [1,3] This enables cascade screening of at-risk relatives and alters surveillance protocols.
3. Aetiology and Pathophysiology
Tumour Origin and Cell Type
Phaeochromocytomas arise from chromaffin cells of the adrenal medulla, which are neuroectodermal in origin, derived from the neural crest. Paragangliomas arise from extra-adrenal chromaffin tissue (paraganglia) located along the sympathetic chain (sympathetic paragangliomas) or parasympathetic nerves (parasympathetic paragangliomas, predominantly in the head and neck). [2]
Anatomical Distribution:
- Adrenal medulla (90%): True phaeochromocytomas
- Organ of Zuckerkandl (5-8%): Sympathetic paraganglia near aortic bifurcation
- Thorax (2%): Mediastinal or cardiac paragangliomas
- Bladder (1%): Bladder wall paragangliomas (cause classic symptoms with micturition)
- Head and neck (3-5%): Carotid body, jugular, vagal paragangliomas (usually parasympathetic; rarely secrete catecholamines)
Molecular Pathophysiology
Recent advances have revealed that phaeochromocytomas and paragangliomas can be classified into molecular clusters based on underlying pathophysiology:
Cluster 1 - Pseudohypoxia Pathway:
- Genes: VHL, SDHA/B/C/D, HIF2A
- Mechanism: Constitutive activation of hypoxia-inducible factors (HIFs) despite normal oxygen
- Associated with high malignancy risk (especially SDHB)
- Tumours are often extra-adrenal and noradrenaline-secreting
Cluster 2 - Kinase Signalling Pathway:
- Genes: RET, NF1, TMEM127, MAX
- Mechanism: Dysregulation of receptor tyrosine kinase and RAS-MAPK signalling
- Lower malignancy risk
- Tumours tend to be adrenal and adrenaline-secreting (especially RET/NF1)
Cluster 3 - Wnt Signalling Pathway:
- Genes: CSDE1, sporadic tumours
- Mechanism: Abnormal Wnt pathway activation
- Mostly sporadic cases
This molecular classification has prognostic and therapeutic implications. [4]
Catecholamine Synthesis and Secretion
Chromaffin cells synthesise catecholamines through the following pathway:
- Tyrosine → (tyrosine hydroxylase) → DOPA (rate-limiting step)
- DOPA → (DOPA decarboxylase) → Dopamine
- Dopamine → (dopamine β-hydroxylase) → Noradrenaline
- Noradrenaline → (phenylethanolamine N-methyltransferase, PNMT) → Adrenaline
Key Points:
- PNMT enzyme (converts noradrenaline → adrenaline) requires cortisol, hence is predominantly active in adrenal medulla (exposed to high cortisol from adjacent cortex)
- Extra-adrenal paragangliomas lack cortisol exposure, thus secrete predominantly noradrenaline or dopamine
- Adrenal phaeochromocytomas can secrete both noradrenaline and adrenaline
- Some tumours (especially malignant paragangliomas) secrete mainly dopamine
Catecholamine Metabolism
Catecholamines are metabolised via two main enzymatic pathways:
-
Catechol-O-methyltransferase (COMT): Converts catecholamines → metanephrines
- Noradrenaline → normetanephrine
- Adrenaline → metanephrine
-
Monoamine oxidase (MAO): Converts catecholamines → vanillylmandelic acid (VMA)
Diagnostic Significance: Metanephrines are produced continuously within the tumour by COMT, independent of catecholamine release. This makes metanephrines far more sensitive markers than catecholamines themselves (which are released episodically). Plasma and urinary metanephrines are thus the biochemical tests of choice. [5,6]
Pathophysiology of Clinical Manifestations
The clinical features of phaeochromocytoma result from excess catecholamine effects on adrenergic receptors:
| Receptor | Catecholamine | Tissue | Effect | Clinical Manifestation |
|---|---|---|---|---|
| Alpha-1 | Noradrenaline >> Adrenaline | Vascular smooth muscle | Vasoconstriction | Hypertension, pallor, cold extremities |
| Beta-1 | Noradrenaline, Adrenaline | Cardiac muscle | Increased heart rate and contractility | Palpitations, tachycardia, arrhythmias |
| Beta-2 | Adrenaline >> Noradrenaline | Vascular smooth muscle (skeletal muscle), bronchi | Vasodilation, bronchodilation | Can cause relative hypotension in adrenaline-secreting tumours |
| Metabolic | Noradrenaline, Adrenaline | Liver, adipose, muscle | Glycogenolysis, lipolysis, gluconeogenesis | Hyperglycaemia, weight loss, sweating |
Paroxysmal vs. Sustained Symptoms:
- Paroxysmal: Episodic catecholamine release causes "attacks" (minutes to hours)
- Sustained: Continuous catecholamine excess causes persistent symptoms
- Approximately 50% have paroxysmal hypertension, 50% sustained hypertension, 5-10% are normotensive
Triggers for Catecholamine Release:
- Physical: Exercise, postural change, abdominal pressure/palpation, micturition (bladder paragangliomas)
- Pharmacological: Metoclopramide, tricyclic antidepressants, sympathomimetics, opioids, glucagon
- Physiological: Stress, pregnancy, anaesthesia/surgery
- Dietary: Tyramine-containing foods (cheese, red wine)
4. Clinical Presentation
Classical Triad
The hallmark presentation is the triad of episodic:
- Headache (80% of patients) - severe, throbbing, generalised
- Sweating (70%) - profuse diaphoresis, often drenching
- Palpitations (70%) - forceful, rapid heartbeat
The presence of all three symptoms in the context of hypertension has a sensitivity of 90% and specificity of 94% for phaeochromocytoma. [2]
Symptoms
Cardinal Symptoms
- Headache (60-90%): Severe, throbbing, generalised; described as "worst headache of my life" during paroxysms
- Excessive sweating (50-70%): Profuse diaphoresis, particularly during paroxysms; may drench clothing
- Palpitations (50-70%): Forceful, rapid, or irregular heartbeat; may be associated with chest discomfort
- Pallor (40-50%): Facial pallor during attacks (NOT flushing, which suggests other diagnosis)
- Anxiety/sense of impending doom (30-40%): Severe psychological distress during paroxysms
Associated Symptoms
- Tremor (20-30%): Fine tremor, particularly of hands
- Nausea and vomiting (20-30%)
- Chest pain (15-20%): Can mimic acute coronary syndrome
- Abdominal pain (10-15%): Vague or localised to tumour
- Weight loss (10-20%): Due to hypermetabolic state
- Weakness and fatigue (variable)
- Visual disturbances (5-10%): Due to hypertensive retinopathy or acute blood pressure changes
Paroxysmal Attacks
"Spells" or "attacks" are characteristic:
- Duration: Seconds to hours (typically 15-60 minutes)
- Frequency: Variable; daily to monthly
- Triggers: Exercise, postural changes, abdominal palpation, certain drugs (see above), emotional stress
- Post-attack: Fatigue, exhaustion; some patients experience reactive hypotension
Signs
General Examination
- Hypertension: Sustained (50-60%), paroxysmal (30%), orthostatic hypotension paradoxically present in 10-20%
- Tachycardia or arrhythmias: Atrial fibrillation, ventricular ectopy
- Pallor: Especially during paroxysms
- Tremor: Fine tremor
- Weight loss: Cachexia in severe cases
Blood Pressure Patterns
- Sustained hypertension: 50-60% of cases; may be severe and resistant to multiple antihypertensives
- Paroxysmal hypertension: 30% of cases; dramatic BP swings (e.g., 240/140 during attacks, 140/90 at baseline)
- Orthostatic hypotension: 10-20%; due to volume depletion and downregulation of alpha receptors
- Normotension: 5-10%; typically dopamine-secreting tumours or subclinical cases
Stigmata of Hereditary Syndromes
- MEN2A/2B:
- Thyroid nodules or surgical scar (medullary thyroid cancer)
- Marfanoid habitus, mucosal neuromas (MEN2B)
- Von Hippel-Lindau:
- Retinal haemangioblastomas (fundoscopy)
- Cerebellar signs (if CNS haemangioblastomas)
- Neurofibromatosis Type 1:
- Café-au-lait spots (≥6 macules > 5mm prepubertal, > 15mm postpubertal)
- Cutaneous/subcutaneous neurofibromas
- Axillary or inguinal freckling (Crowe's sign)
- Lisch nodules (iris hamartomas on slit-lamp)
Cardiac Manifestations
- Catecholamine-induced cardiomyopathy: Heart failure symptoms (dyspnoea, orthopnoea, peripheral oedema) in severe chronic cases [18]
- Arrhythmias: Atrial fibrillation, ventricular ectopy, rarely ventricular tachycardia [19]
- Acute coronary syndrome: Chest pain mimicking myocardial infarction due to coronary vasospasm or demand ischaemia
Red Flags
[!CAUTION] IMMEDIATE CONCERN - Phaeochromocytoma Crisis
- Severe hypertension (BP > 220/120 mmHg) with end-organ damage
- Hypertensive encephalopathy (confusion, seizures, altered consciousness)
- Acute pulmonary oedema or myocardial infarction
- Stroke or intracranial haemorrhage
- Hypertensive crisis precipitated by surgery, anaesthesia, or certain drugs
Action: Immediate ICU admission, IV phentolamine or nitroprusside, urgent endocrine consult
[!WARNING] High Clinical Suspicion Warranted
- Resistant hypertension (requiring ≥3 antihypertensives)
- Paroxysmal symptoms (especially triad of headache, sweating, palpitations)
- Hypertension in young patient (less than 40 years, especially less than 30)
- Hypertension with adrenal mass on imaging
- Family history of phaeochromocytoma, MEN2, VHL, or SDH mutations
- Personal history of other features of hereditary syndrome
- Paradoxical hypertensive response to anaesthesia or antihypertensives (especially beta-blockers)
- Incidental adrenal mass ("adrenal incidentaloma")
5. Clinical Examination
Structured Approach to Examination
General Inspection
- Habitus: Weight loss, cachexia (chronic catecholamine excess); Marfanoid features (MEN2B)
- Skin:
- Café-au-lait spots (≥6; NF1)
- Neurofibromas (NF1)
- Mucosal neuromas - lips, tongue (MEN2B)
- Diaphoresis (sweating)
- Pallor (not flushing)
- Behaviour: Anxiety, restlessness
Vital Signs
- Blood pressure: ESSENTIAL - measure in both arms, supine and standing (orthostatic changes)
- Document BP during asymptomatic period and during/immediately after "attack" if possible
- Heart rate: Tachycardia, irregular (arrhythmias)
- Respiratory rate: May be elevated if pulmonary oedema
- Temperature: Usually normal
Cardiovascular
- Pulses: Bounding pulse (high stroke volume), tachycardia
- Heart sounds: May reveal arrhythmias; third heart sound (S3) if heart failure
- Signs of heart failure: Raised JVP, peripheral oedema, pulmonary crepitations (catecholamine cardiomyopathy)
Abdominal Examination
- Inspection: Surgical scars (previous adrenal surgery)
- Palpation: Rarely, large phaeochromocytomas may be palpable (avoid vigorous palpation - can precipitate crisis)
- Renal bruits: Listen for bruits (consider renovascular hypertension in differential)
Neurological
- Fundoscopy: Hypertensive retinopathy (arteriovenous nipping, flame haemorrhages, papilloedema)
- Cerebellar signs: If VHL disease with cerebellar haemangioblastomas
- Peripheral neuropathy: May occur in NF1
Endocrine Stigmata
- Neck examination: Thyroid nodules or thyroidectomy scar (MEN2 - medullary thyroid cancer)
- Ophthalmology: Retinal haemangioblastomas (VHL - require specialist fundoscopy/fluorescein angiography)
- Axillary/inguinal freckling: Crowe's sign (NF1)
6. Differential Diagnosis
Phaeochromocytoma is rare and can mimic many common conditions. The differential diagnosis is broad:
Primary Differentials
| Condition | Distinguishing Features | Key Investigations |
|---|---|---|
| Essential hypertension | Gradual onset, no paroxysms, no triad, family history | Normal metanephrines |
| Anxiety/panic disorder | Psychological triggers, responds to anxiolytics, younger patients | Normal metanephrines; psychiatric assessment |
| Thyrotoxicosis | Heat intolerance, weight loss despite appetite, tremor, goitre, exophthalmos | Elevated free T4/T3, suppressed TSH |
| Hypoglycaemia | Episodic, relieved by food, low glucose during attack | Glucose less than 3.0 mmol/L during symptoms |
| Carcinoid syndrome | Flushing (not pallor), diarrhoea, bronchospasm, right-sided valvular disease | Elevated 24h urinary 5-HIAA |
| Menopause | Age-appropriate (perimenopausal women), hot flushes, amenorrhoea | Elevated FSH; normal metanephrines |
| Mastocytosis | Flushing, pruritus, urticaria, anaphylaxis, diarrhoea | Elevated serum tryptase, urinary methylhistamine |
| Cocaine/amphetamine use | Relevant history, positive toxicology, psychiatric features | Urine drug screen |
Important "Must-Not-Miss" Differentials
| Condition | Why Important | Distinguishing Features |
|---|---|---|
| Renovascular hypertension | Treatable cause of secondary hypertension | Renal artery bruit, asymmetric kidneys on imaging, elevated renin |
| Cushing's syndrome | Curable endocrine hypertension | Central obesity, striae, proximal myopathy; elevated 24h urinary free cortisol |
| Primary hyperaldosteronism (Conn's) | Most common endocrine cause of hypertension (5-10%) | Hypokalaemia, metabolic alkalosis; elevated aldosterone:renin ratio |
| Aortic coarctation | Surgically correctable | Radio-femoral delay, upper limb hypertension, rib notching on CXR |
| Intracranial lesion (tumour, haemorrhage) | Can cause secondary hypertension and headaches | Focal neurology, papilloedema; CT/MRI brain |
Rarer Mimics
- Baroreflex failure: After neck surgery/radiation; extreme BP lability
- Autonomic dysfunction: Postural hypotension predominant
- Factitious disorder: Self-administration of sympathomimetics
- Paroxysmal atrial tachycardia: Palpitations without hypertension/sweating triad
7. Investigations
Diagnostic Approach
The diagnosis of phaeochromocytoma follows a two-step process:
- Biochemical confirmation of excess catecholamine production
- Anatomical localisation of the tumour with imaging
Step 1: Biochemical Confirmation
First-Line Biochemical Tests
Plasma Free Metanephrines (GOLD STANDARD):
- Sensitivity: 96-99%
- Specificity: 89-95%
- Method: Blood sample after 20-30 minutes supine rest (reduces false positives from stress/postural changes)
- Interpretation:
- "Normal: Excludes phaeochromocytoma with > 95% certainty (excellent negative predictive value)"
-
2× upper limit of normal: Highly suggestive of phaeochromocytoma
- 1-2× upper limit: Grey zone; may require repeat testing or clonidine suppression test
- Components: Normetanephrine (from noradrenaline) and metanephrine (from adrenaline)
- Evidence: Multiple meta-analyses confirm superiority over other tests [5,6]
24-Hour Urinary Fractionated Metanephrines (ALTERNATIVE):
- Sensitivity: 92-97%
- Specificity: 91-98%
- Advantages: Less affected by stress; integrates catecholamine production over 24 hours
- Disadvantages: Requires complete collection; patient compliance
- Interpretation: > 2× upper limit highly suggestive
| Test | Sensitivity | Specificity | Advantages | Disadvantages |
|---|---|---|---|---|
| Plasma free metanephrines | 96-99% | 89-95% | Single sample, highest sensitivity | Affected by stress/posture; false positives |
| 24h urinary metanephrines | 92-97% | 91-98% | Less stress effect, integrates 24h | Requires complete collection |
| Plasma catecholamines | 84-92% | 90-95% | - | Episodic secretion; many false negatives |
| Urinary VMA | 64-86% | 95-98% | - | Lowest sensitivity; outdated |
Factors Causing False-Positive Results
Multiple medications and conditions can elevate metanephrines:
Medications (withhold ≥2 weeks before testing if possible):
- Tricyclic antidepressants
- Phenoxybenzamine
- Labetalol, sotalol (can elevate normetanephrine)
- Sympathomimetics (decongestants, cocaine, amphetamines)
- Paracetamol (acetaminophen) - interferes with HPLC assays
- Monoamine oxidase inhibitors (MAOIs)
Physiological States:
- Acute stress, pain, severe illness
- Obstructive sleep apnoea
- Withdrawal from alcohol or clonidine
Medical Conditions:
- Renal failure (impaired clearance)
- Obstructive sleep apnoea
- Heart failure
Confirmatory Tests (if Equivocal Results)
Clonidine Suppression Test:
- Principle: Clonidine (central alpha-2 agonist) suppresses catecholamine release from normal adrenal medulla but NOT from autonomous phaeochromocytoma
- Method: Measure baseline plasma catecholamines, give clonidine 0.3 mg orally, repeat plasma catecholamines at 3 hours
- Interpretation: Failure to suppress plasma noradrenaline by > 50% or to less than 500 pg/mL suggests phaeochromocytoma
- Use: Reserved for borderline biochemical results
Step 2: Anatomical Localisation
Once biochemical diagnosis is confirmed, imaging localises the tumour.
First-Line Imaging
CT Adrenals with Contrast:
- Sensitivity: 93-100% for adrenal phaeochromocytomas
- Protocol: Thin-slice (2-3 mm) CT with arterial and venous phases
- Findings:
- Well-defined, hypervascular mass (enhances avidly)
- Median size 3-5 cm (range 1-15 cm)
- Heterogeneous (haemorrhage, necrosis, cystic change common)
-
10 Hounsfield units (HU) on unenhanced CT (distinguishes from lipid-rich adenoma)
- Advantages: Widely available, fast, excellent spatial resolution
- Disadvantages: Radiation, iodinated contrast (risk of precipitating crisis - ensure adequate alpha-blockade first)
MRI Adrenals:
- Sensitivity: 90-100%
- Sequences: T1, T2, chemical shift imaging
- Findings:
- Hyperintense ("light bulb sign") on T2-weighted imaging (classic but not universal)
- Does NOT drop signal on opposed-phase chemical shift (vs. adenomas which do)
- Advantages: No radiation, no iodinated contrast, better for extra-adrenal paragangliomas, pregnancy
- Disadvantages: Longer acquisition, less available, expensive
Second-Line/Functional Imaging
Reserved for specific scenarios:
- Extra-adrenal/metastatic disease suspected
- CT/MRI negative despite positive biochemistry
- Hereditary syndromes (multifocal disease)
- Preoperative mapping
123I-MIBG Scintigraphy:
- Principle: MIBG (meta-iodobenzylguanidine) is a noradrenaline analogue taken up by chromaffin cells
- Sensitivity: 77-90% (lower for extra-adrenal, SDHB-mutated, malignant tumours)
- Specificity: 95-100%
- Protocol: Images at 24 and 48 hours post-injection
- Use: Confirms functional tissue; helpful for identifying metastases
- Limitations: Lower sensitivity than PET; false negatives in dedifferentiated tumours
68Ga-DOTATATE PET/CT (Somatostatin Receptor Imaging):
- Principle: Most phaeochromocytomas/paragangliomas express somatostatin receptors (especially SSTR2)
- Sensitivity: 90-100% (superior to MIBG, especially for SDH-mutated and metastatic disease) [7]
- Advantages: Excellent for SDHx-related tumours, head/neck paragangliomas, metastatic disease
- Availability: Increasingly available; superior to MIBG for paragangliomas [20]
- Use: Now first-line functional imaging in many centres; essential for SDHB-mutated cases
18F-FDG PET/CT:
- Use: Detects metastatic disease, especially in aggressive/malignant tumours with high metabolic activity
- Sensitivity: Variable (60-90% for metastases)
- Indications: SDHB-mutated tumours (high malignancy risk), staging metastatic disease
| Imaging Modality | Sensitivity | Best Use | Limitations |
|---|---|---|---|
| CT adrenals | 93-100% | First-line adrenal localisation | Radiation; contrast risk |
| MRI adrenals | 90-100% | Pregnancy, extra-adrenal, contrast allergy | Longer scan; expensive |
| 123I-MIBG | 77-90% | Functional confirmation, metastases | Lower sensitivity for paragangliomas, SDHx |
| 68Ga-DOTATATE PET | 90-100% | SDHx tumours, paragangliomas, metastases | Availability; cost |
| 18F-FDG PET | 60-90% | Metastatic/aggressive disease | Lower specificity |
Step 3: Genetic Testing
Genetic testing is now recommended for ALL patients with phaeochromocytoma or paraganglioma, regardless of age, family history, or tumour location. [1,3]
Genes to Test (in order of prevalence):
- SDHB, SDHD, SDHC: Succinate dehydrogenase subunits (most common in paragangliomas)
- VHL: Von Hippel-Lindau
- RET: MEN2A/2B
- NF1: Neurofibromatosis type 1
- TMEM127, MAX, SDHA, SDHAF2: Rarer genes
Clinical Utility:
- Identifies hereditary syndrome (enables cascade screening of relatives)
- Predicts malignancy risk (SDHB high risk)
- Guides surveillance (e.g., bilateral adrenal disease in MEN2)
- Influences surgical planning
Timing: Can be performed concurrently with biochemical/imaging workup; ideally before surgery to guide extent of resection.
Additional Investigations
Routine Laboratory Tests:
- FBC: Haemoconcentration (volume depletion from chronic vasoconstriction)
- U&Es: Hypokalaemia (rare; from chronic catecholamine excess)
- Glucose: Hyperglycaemia (catecholamine-induced insulin resistance)
- Calcium: Hypercalcaemia if MEN2A (concomitant hyperparathyroidism)
Cardiovascular Workup:
- ECG: LVH, arrhythmias, ischaemic changes
- Echocardiography: Left ventricular hypertrophy, assess for catecholamine-induced cardiomyopathy (reduced ejection fraction), valvular disease
- 24h BP monitoring: Document BP variability, paroxysmal hypertension
Pre-operative Assessment:
- Comprehensive cardiac evaluation
- Anaesthetic review (high-risk surgery)
8. Management
The management of phaeochromocytoma requires a multidisciplinary approach involving endocrinology, surgery, anaesthetics, and clinical genetics. The key principle is safe preoperative preparation followed by surgical resection.
Overview of Management Pathway
- Biochemical confirmation → Plasma/urinary metanephrines
- Imaging localisation → CT/MRI ± functional imaging
- Genetic testing → All patients
- Preoperative preparation → Alpha-blockade, then beta-blockade, volume expansion
- Surgical resection → Laparoscopic adrenalectomy (preferred)
- Postoperative management → Monitoring for hypotension/hypoglycaemia
- Lifelong follow-up → Annual biochemical screening for recurrence
Preoperative Medical Preparation (CRITICAL)
Adequate preoperative alpha-adrenergic blockade is ESSENTIAL to prevent intraoperative hypertensive crisis, arrhythmias, and cardiovascular complications. [1,8] Inadequate preparation is associated with significant perioperative morbidity and mortality.
Step 1: Alpha-Adrenergic Blockade (FIRST)
Phenoxybenzamine (non-selective, irreversible alpha-blocker):
- Regimen: Start at 10 mg BD, increase by 10-20 mg every 2-3 days
- Target dose: 20-40 mg BD (or up to 1 mg/kg/day in divided doses)
- Duration: Minimum 10-14 days preoperatively (often 2-4 weeks)
- Endpoints:
- Normotension or mild orthostatic hypotension (systolic drop > 10 mmHg on standing)
- Resolution of symptoms (headaches, sweating, palpitations)
- Blood pressure less than 130/80 mmHg seated; orthostatic systolic drop ≥10 mmHg acceptable
- Side effects: Orthostatic hypotension, nasal congestion, fatigue, reflex tachycardia
Doxazosin (selective alpha-1 blocker, reversible):
- Regimen: Start 2-4 mg OD, titrate up to 8-16 mg OD (maximum 32 mg/day)
- Advantages: Once-daily dosing, better tolerated, reversible
- Evidence: Several studies show comparable efficacy to phenoxybenzamine [8]
- Growing preference: Increasingly used as first-line in many centres
Comparison:
| Feature | Phenoxybenzamine | Doxazosin |
|---|---|---|
| Selectivity | Non-selective (α1 + α2) | Selective α1 |
| Binding | Irreversible | Reversible |
| Dosing | BD (twice daily) | OD (once daily) |
| Side effects | More orthostatic hypotension | Better tolerated |
| Evidence | Traditional gold standard | Comparable outcomes [8] |
Step 2: Volume Expansion
- Rationale: Chronic catecholamine excess causes vasoconstriction → reduced plasma volume. Alpha-blockade causes vasodilation, and without volume expansion, severe hypotension can occur.
- Methods:
- High-salt diet (> 5 g/day sodium)
- Oral fluids (2-3 L/day)
- IV fluids if necessary (especially day before surgery)
- Monitoring: Aim for resolution of orthostatic hypotension
Step 3: Beta-Adrenergic Blockade (ONLY AFTER adequate alpha-blockade)
CRITICAL: NEVER give beta-blocker before or without alpha-blockade. Beta-blockade alone blocks beta-2 mediated vasodilation, leaving alpha-mediated vasoconstriction unopposed → severe hypertensive crisis.
Indications:
- Persistent tachycardia (HR > 100 bpm) despite alpha-blockade
- Arrhythmias (e.g., atrial fibrillation)
Agents:
- Propranolol: 20-40 mg TDS (non-selective)
- Atenolol: 25-50 mg OD (selective beta-1)
- Labetalol is sometimes used (combined alpha/beta blocker), but has predominantly beta-blocking activity and should still only be used after adequate alpha-blockade
Timing: Start 2-3 days after achieving adequate alpha-blockade
Step 4: Additional Medications
Calcium Channel Blockers (e.g., amlodipine, nifedipine):
- Used as adjunct if inadequate BP control despite alpha-blockade
- Useful in patients intolerant of alpha-blockers
Metyrosine (Alpha-Methyl-Para-Tyrosine):
- Mechanism: Inhibits tyrosine hydroxylase (rate-limiting enzyme in catecholamine synthesis)
- Use: Reserved for severe cases, metastatic disease, or patients intolerant of alpha-blockers
- Dose: 250 mg QDS, titrated to 1-4 g/day
- Side effects: Sedation, diarrhoea, crystalluria
- Availability: Limited; expensive
Preoperative Assessment Checklist
Before proceeding to surgery, confirm:
- ✅ Blood pressure less than 130/80 mmHg seated
- ✅ Orthostatic hypotension present (systolic drop ≥10 mmHg)
- ✅ No paroxysmal symptoms (headache, sweating, palpitations) for ≥2 weeks
- ✅ Heart rate less than 100 bpm
- ✅ No arrhythmias on ECG/24h monitoring
- ✅ Adequate volume status (no haemoconcentration)
Surgical Management
Indications for Surgery
Surgery is the definitive treatment and is indicated in:
- All localised phaeochromocytomas (benign or potentially malignant)
- Resectable metastatic disease (debulking)
Contraindications (relative):
- Extensive unresectable metastatic disease (consider medical management/MIBG therapy)
- Severe comorbidities precluding surgery
Surgical Approach
Laparoscopic Adrenalectomy (PREFERRED):
- Advantages: Minimally invasive, less pain, shorter recovery, reduced complications
- Evidence: Multiple RCTs and meta-analyses demonstrate comparable outcomes to open surgery with fewer complications [11]
- Approach: Transabdominal or retroperitoneal
- Success rate: > 95% for tumours less than 6 cm
Open Adrenalectomy:
- Indications: Large tumours (> 6 cm), invasive tumours, suspected malignancy, extensive local involvement
- Approach: Anterior midline or subcostal
Key Surgical Principles:
- Minimal tumour manipulation: Avoid excessive handling to prevent catecholamine surge
- Early venous ligation: Ligate adrenal vein early to prevent catecholamine release into systemic circulation
- Careful dissection: Tumours can be friable and vascular
Intraoperative Management
Requires expert anaesthetic management:
Monitoring:
- Arterial line (continuous BP monitoring)
- Central venous line (volume status, vasopressor administration)
- ECG (arrhythmia detection)
BP Management:
- Hypertensive crises (during tumour manipulation):
- "IV phentolamine (alpha-blocker): 1-5 mg boluses"
- "IV sodium nitroprusside infusion: 0.5-10 mcg/kg/min"
- "IV magnesium sulphate: 2-4 g bolus (also anti-arrhythmic)"
- Hypotension (post-tumour resection - very common):
- "IV fluids: Large-volume crystalloid"
- "Vasopressors if needed: noradrenaline infusion"
Arrhythmia Management:
- Ventricular arrhythmias: Lidocaine, magnesium, beta-blockers
- Avoid drugs that trigger catecholamine release (e.g., droperidol)
Bilateral Adrenalectomy
- Indications: Bilateral phaeochromocytomas (e.g., MEN2, VHL)
- Consequence: Lifelong adrenal insufficiency → requires lifelong glucocorticoid and mineralocorticoid replacement
- Staged vs. Simultaneous: Usually staged (3-6 months apart) to minimise risk, unless urgent
Postoperative Management
Immediate Postoperative Period (ICU/HDU)
Monitoring:
- Continuous BP and ECG monitoring (first 24-48 hours)
- Blood glucose monitoring
Common Issues:
- Hypotension (most common):
- "Mechanism: Loss of catecholamine drive + residual alpha-blockade + volume shifts"
- "Management: IV fluids (often 3-5 L required); vasopressors if refractory"
- Hypoglycaemia:
- "Mechanism: Loss of catecholamine-mediated glycogenolysis, residual beta-cell suppression recovering"
- "Management: Regular glucose monitoring; IV dextrose if needed"
- Acute adrenal insufficiency (if bilateral adrenalectomy):
- "Management: Hydrocortisone 100 mg IV TDS initially, then oral replacement"
Biochemical Confirmation of Cure
- Plasma metanephrines at 2-4 weeks postoperatively
- Should normalise if complete resection
- Persistently elevated → residual tumour or metastases
Management of Malignant/Metastatic Phaeochromocytoma
Malignancy is defined by the presence of metastases in non-chromaffin tissue (lymph nodes, liver, lung, bone). There are no reliable histological features to predict malignancy. [10]
Systemic Treatment Options
131I-MIBG Radiotherapy:
- Indication: Metastatic disease with MIBG-avid tumours
- Mechanism: Beta-emitting radioisotope selectively taken up by chromaffin cells
- Efficacy: Partial response or disease stabilisation in 30-50%; symptom improvement in > 70%
- Side effects: Myelosuppression, hypothyroidism (requires thyroid blockade)
Peptide Receptor Radionuclide Therapy (PRRT) with 177Lu-DOTATATE:
- Indication: Metastatic disease with DOTATATE-avid tumours (somatostatin receptor-positive)
- Evidence: Emerging; promising results in neuroendocrine tumours
- Availability: Specialist centres
Chemotherapy:
- CVD regimen: Cyclophosphamide, vincristine, dacarbazine
- Response rate: 30-50% (usually partial, temporary)
- Use: Reserved for progressive metastatic disease
Targeted Therapy:
- Sunitinib (multi-kinase inhibitor): Recent FIRSTMAPPP trial showed progression-free survival benefit [10]
- Other agents: Cabozantinib, lenvatinib under investigation
Surgery:
- Debulking surgery for symptomatic metastases or dominant masses
Long-Term Follow-Up
ALL patients require lifelong follow-up due to risk of:
- Recurrence: 5-15% over 10 years (higher in hereditary syndromes, malignant disease)
- Development of new primary tumours: Especially in hereditary syndromes (e.g., bilateral disease in MEN2)
Follow-Up Protocol
Annual Biochemical Screening:
- Plasma or 24h urinary fractionated metanephrines
- Lifelong (recurrence can occur decades later)
Imaging:
- If biochemistry abnormal → repeat CT/MRI
- Consider routine imaging every 1-2 years in high-risk patients (SDHB mutations, malignant disease)
Genetic Counselling and Cascade Screening:
- If germline mutation identified → refer first-degree relatives for genetic testing
- Annual biochemical screening for mutation carriers (even if asymptomatic)
Special Populations:
- MEN2: Annual screening from age 8-10; consider prophylactic thyroidectomy
- VHL: MRI brain/spine for haemangioblastomas; renal imaging for RCC
- SDHB: Aggressive imaging surveillance due to high malignancy risk
9. Complications
Complications of Untreated Phaeochromocytoma
| Complication | Mechanism | Frequency | Clinical Significance |
|---|---|---|---|
| Hypertensive crisis | Catecholamine surge | 5-10% present this way | Life-threatening; stroke, MI, pulmonary oedema |
| Catecholamine cardiomyopathy | Chronic beta-1 stimulation, myocardial toxicity | 10-15% [18] | Reversible if treated; can progress to heart failure |
| Stroke | Severe hypertension, cerebral vasospasm | 2-5% | Haemorrhagic or ischaemic |
| Myocardial infarction | Coronary vasospasm, demand ischaemia | 2-5% | Can occur with normal coronary arteries |
| Arrhythmias | Catecholamine excess [19] | 20-30% | AF most common; VT rare but life-threatening |
| Pregnancy complications | Hypertensive crisis during labour | Variable | Maternal and fetal mortality if undiagnosed |
Perioperative Complications
| Complication | Timing | Frequency | Prevention | Management |
|---|---|---|---|---|
| Intraoperative hypertensive crisis | During tumour manipulation | 20-40% | Adequate alpha-blockade | IV phentolamine, nitroprusside |
| Postoperative hypotension | First 24-48h | 30-50% | Volume expansion preop | IV fluids, vasopressors if needed |
| Hypoglycaemia | First 24h | 5-10% | Monitor glucose | IV dextrose |
| Arrhythmias | Intraoperative | 10-20% | Adequate preop prep | Lidocaine, beta-blockers, magnesium |
| Pneumothorax | Intraoperative (if thoracic approach) | less than 5% | Careful surgical technique | Chest drain |
Long-Term Complications
- Recurrence: 5-15% at 10 years; lifelong surveillance required
- Metachronous tumours: In hereditary syndromes (e.g., bilateral phaeochromocytomas)
- Adrenal insufficiency: If bilateral adrenalectomy → lifelong replacement therapy
10. Prognosis
Overall Outcomes
| Outcome Measure | Rate | Notes |
|---|---|---|
| Cure with surgery | 90% | If complete resection of benign tumour [11] |
| Recurrence | 5-15% at 10 years | Higher in hereditary syndromes, extra-adrenal, large tumours |
| Malignancy | 10-17% | Defined by presence of metastases [10] |
| 5-year survival (malignant) | 40-60% | Depends on mutation, metastatic burden |
Prognostic Factors
Favourable:
- Adrenal location (vs. extra-adrenal)
- Sporadic (vs. SDHB mutation)
- Completely resected
- Small tumour (less than 5 cm)
Unfavourable:
- SDHB mutation (30-70% malignancy risk) [10]
- Extra-adrenal (paragangliomas have higher malignancy rates)
- Large tumour (> 5 cm)
- Extensive local invasion
- High proliferative index (Ki-67 > 3%)
- Young age at diagnosis (less than 40 years) in some studies
Genotype-Specific Prognosis
| Mutation | Malignancy Risk | Recurrence Risk | Special Considerations |
|---|---|---|---|
| Sporadic | 10% | 5-10% | Standard surveillance |
| SDHB | 30-70% | High | Aggressive surveillance; consider PRRT |
| MEN2 | less than 5% | High (bilateral) | Screen for MTC, hyperparathyroidism |
| VHL | 5% | Moderate | Screen for CNS haemangioblastomas, RCC |
| NF1 | less than 5% | Low | Usually benign |
Quality of Life
- Post-curative surgery: Excellent; most patients become normotensive or require fewer antihypertensives
- Catecholamine cardiomyopathy: Often reversible if treated early [18]
- Lifelong surveillance: Required; annual biochemistry; impacts QoL
11. Evidence and Guidelines
Key Guidelines
-
Lenders JWM et al. (2014) — Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2014;99(6):1915-1942. PMID: 24893135 | DOI: 10.1210/jc.2014-1498
- Comprehensive guideline covering diagnosis, imaging, genetic testing, preoperative management, and surveillance
- Recommends plasma free metanephrines as first-line test
- Advocates genetic testing for all patients
-
Fishbein L et al. (2021) — The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma. Pancreas 2021;50(4):469-493. PMID: 33939658 | DOI: 10.1097/MPA.0000000000001792
- Specific guidance on malignant/metastatic disease
- Discusses PRRT, MIBG therapy, systemic treatments
-
Taïeb D et al. (2023) — Clinical consensus guideline on the management of phaeochromocytoma and paraganglioma in patients harbouring germline SDHD pathogenic variants. Lancet Diabetes Endocrinol 2023;11(5):345-361. PMID: 37011647 | DOI: 10.1016/S2213-8587(23)00038-4
- Specific guidance for SDH-mutated tumours
- Emphasises high malignancy risk in SDHB
Landmark Studies
-
Neumann HPH et al. (2019) — Pheochromocytoma. N Engl J Med 2019;381(6):552-565. PMID: 31390501
- Comprehensive review of pathophysiology, genetics, diagnosis, and management
-
Chen Y et al. (2017) — Accuracy of Plasma Free Metanephrines in the Diagnosis of Pheochromocytoma and Paraganglioma: A Systematic Review and Meta-Analysis. Endocr Pract 2017;23(10):1169-1177. PMID: 28704098 | DOI: 10.4158/EP171877.OR
- Meta-analysis confirming 96% sensitivity and 92% specificity of plasma free metanephrines
-
Därr R et al. (2017) — Accuracy of recommended sampling and assay methods for the determination of plasma-free and urinary fractionated metanephrines in the diagnosis of pheochromocytoma and paraganglioma: a systematic review. Endocrine 2017;56(3):495-503. PMID: 28405881 | DOI: 10.1007/s12020-017-1300-y
- Systematic review on optimal biochemical testing methods
-
Naji M et al. (2012) — 68Ga-labelled peptides in the management of neuroectodermal tumours. Eur J Nucl Med Mol Imaging 2012;39 Suppl 1:S61-7. PMID: 22388623 | DOI: 10.1007/s00259-011-1990-y
- Discusses role of 68Ga-DOTATATE PET in phaeochromocytoma/paraganglioma
-
Zhu CY et al. (2022) — Comparison of Preoperative Alpha-blockade for Resection of Paraganglioma and Pheochromocytoma. Endocr Pract 2022;28(9):873-879. PMID: 35809774 | DOI: 10.1016/j.eprac.2022.06.013
- Comparative effectiveness of phenoxybenzamine vs. doxazosin
-
Farrugia FA et al. (2011) — Pheochromocytoma. Endocr Regul 2011;45(2):97-104.
- Epidemiology and clinical features
-
Baudin E et al. (2024) — Sunitinib for metastatic progressive phaeochromocytomas and paragangliomas: results from FIRSTMAPPP, an academic, multicentre, international, randomised, placebo-controlled, double-blind, phase 2 trial. Lancet 2024;403(10431):1103-1112. PMID: 38402886 | DOI: 10.1016/S0140-6736(23)02554-0
- RCT demonstrating efficacy of sunitinib in metastatic disease
- Ma W et al. (2020) — Surgical outcomes of a randomized controlled trial compared robotic versus laparoscopic adrenalectomy for pheochromocytoma. Eur J Surg Oncol 2020;46(10 Pt A):1843-1848. PMID: 32723609 | DOI: 10.1016/j.ejso.2020.04.001
- RCT comparing surgical approaches
Additional Key References
- Leonard JB et al. (2018) — Metoclopramide induced pheochromocytoma crisis. Am J Emerg Med 2018;36(6):1124.e1-1124.e2. PMID: 29534916 | DOI: 10.1016/j.ajem.2018.03.009
- Case report of drug-induced crisis
- Hu J et al. (2024) — Evaluation of Adrenal Incidentaloma. Surg Clin North Am 2024;104(4):831-848. PMID: 38944503 | DOI: 10.1016/j.suc.2024.02.012
- Review of adrenal incidentaloma workup including phaeochromocytoma screening
- Reisch N et al. (2006) — Pheochromocytoma: presentation, diagnosis and treatment. J Hypertens 2006;24(12):2331-9. PMID: 17082709
- Classic review of clinical presentation
- Gild ML et al. (2025) — Pheochromocytoma in MEN2. Recent Results Cancer Res 2025 (epub ahead of print). PMID: 40102259 | DOI: 10.1007/978-3-031-80396-3_8
- MEN2-specific phaeochromocytoma management
- Lefebvre M et al. (2014) — Pheochromocytoma and paraganglioma syndromes: genetics and management update. Curr Oncol 2014;21(1):e8-e17. PMID: 24523625 | DOI: 10.3747/co.21.1579
- Genetic syndromes update
- Raygada M et al. (2011) — Hereditary paragangliomas. Adv Otorhinolaryngol 2011;70:99-106. PMID: 21358191 | DOI: 10.1159/000322484
- Review of hereditary paraganglioma syndromes
- Kumar A et al. (2021) — Catecholamine-induced cardiomyopathy: an endocrinologist's perspective. Rev Cardiovasc Med 2021;22(4):1277-1291. PMID: 34957765 | DOI: 10.31083/j.rcm2204130
- Review of cardiac complications
- Nazari MA et al. (2020) — Pathophysiology and Acute Management of Tachyarrhythmias in Pheochromocytoma: JACC Review Topic of the Week. J Am Coll Cardiol 2020;76(4):451-464. PMID: 32703516 | DOI: 10.1016/j.jacc.2020.04.080
- Cardiac arrhythmias in phaeochromocytoma
- Dodamani MH et al. (2022) — Comparison of the Sensitivity of 68Ga-DOTATATE PET/CT with Other Imaging Modalities in Detecting Head and Neck Paraganglioma: Experience from Western India. World J Nucl Med 2022;21(3):234-241. PMID: 36060084 | DOI: 10.1055/s-0042-1751030
- Comparative imaging study demonstrating superiority of DOTATATE PET
12. Patient Explanation
What is Phaeochromocytoma?
Phaeochromocytoma (pronounced "fee-oh-crow-moh-sy-TOH-mah") is a rare tumour of the adrenal gland—a small gland that sits on top of each kidney. This tumour produces too much adrenaline (the "fight-or-flight" hormone), causing high blood pressure, severe headaches, sweating, and a fast heartbeat.
Most phaeochromocytomas are benign (not cancer), but they can be dangerous if not treated because the sudden surges of adrenaline can cause very high blood pressure, which can lead to stroke, heart attack, or other serious problems.
What Are the Symptoms?
The most common symptoms are:
- Severe headaches (often described as the worst headache you've ever had)
- Heavy sweating (drenching sweats, sometimes at night)
- Fast or forceful heartbeat (palpitations)
- High blood pressure (sometimes very high, or suddenly spiking)
- Feeling anxious or panicky
- Pale skin (not flushing or redness)
These symptoms can come in "attacks" or "spells" lasting minutes to hours, or they can be present all the time.
How is it Diagnosed?
- Blood or urine test: Measures hormones called "metanephrines" that the tumour produces. This test is very accurate.
- Scan (CT or MRI): Locates the tumour on the adrenal gland or elsewhere in the body.
- Genetic test: Checks if the condition runs in your family (about 30-40% of cases are hereditary).
How is it Treated?
The main treatment is surgery to remove the tumour. However, before surgery, you will need to take medications for at least 2 weeks to prepare your body:
- Alpha-blocker medication (e.g., phenoxybenzamine or doxazosin): This lowers your blood pressure and prevents dangerous spikes during surgery. You must take this FIRST.
- Beta-blocker medication (if needed): This slows your heart rate. It is only given AFTER the alpha-blocker, never before.
- Extra salt and fluids: You'll be asked to eat more salt and drink plenty of water to expand your blood volume.
Once you're ready, the surgeon will remove the tumour, usually using keyhole (laparoscopic) surgery. After surgery, your blood pressure and symptoms should improve dramatically.
What Happens After Surgery?
- Most people are cured after surgery.
- You will need a follow-up blood test a few weeks after surgery to confirm the tumour is gone.
- You will need lifelong annual blood tests to check that the tumour hasn't come back (this is rare but possible).
- If the condition runs in your family, your relatives may need to be tested.
What if it Runs in My Family?
If genetic testing finds a hereditary cause, your doctor will:
- Recommend that your close relatives (parents, siblings, children) get tested.
- Arrange regular check-ups for you to monitor for new tumours.
- Screen for other conditions associated with the genetic syndrome.
Key Takeaway
Phaeochromocytoma is a rare but treatable condition. With proper preparation and surgery, most people are cured and their symptoms resolve. Lifelong follow-up is important to ensure it doesn't come back.
13. Examination Focus (MRCP/FRACP Viva)
Viva Opening Statement (30 seconds)
"Phaeochromocytoma is a rare catecholamine-secreting tumour of the adrenal medulla, characterised by the classical triad of episodic headache, sweating, and palpitations with hypertension. Diagnosis is by plasma or urinary metanephrines—the gold standard test with 96% sensitivity. Up to 40% are hereditary, so genetic testing is essential for all patients. Management requires meticulous preoperative alpha-blockade followed by beta-blockade—crucially, NEVER beta-block first—before surgical resection, which is curative in 90%."
Key Facts for Viva
Must-Know Points:
- Classical triad: Headache (80%), sweating (70%), palpitations (70%)
- "5 Ps": Pressure, Pain, Perspiration, Palpitations, Pallor
- Biochemical diagnosis: Plasma free metanephrines (sensitivity 96-99%, specificity 89-95%) [5,6]
- Rule of 10s (updated): 10% extra-adrenal, 10% bilateral, 10-17% malignant, 40% hereditary (not 10%)
- Genetic syndromes: MEN2 (RET), VHL, NF1, SDH mutations (SDHB highest malignancy risk)
- Preoperative preparation: Alpha-blockade FIRST (phenoxybenzamine or doxazosin), then beta-blockade
- Imaging: CT/MRI adrenals first-line; 68Ga-DOTATATE PET for paragangliomas/SDH-mutated tumours
- Malignancy: Defined by metastases; 30-70% in SDHB mutations [10]
- Lifelong follow-up: Annual metanephrines; recurrence risk 5-15%
Common Viva Questions and Model Answers
Q1: How would you investigate a patient with suspected phaeochromocytoma?
"I would follow a two-step approach: biochemical confirmation, then anatomical localisation. First, I'd measure plasma free metanephrines after 20 minutes supine rest—this is the gold standard test with 96% sensitivity. Levels more than twice the upper limit are highly suggestive. Alternatively, 24-hour urinary fractionated metanephrines can be used. Once biochemistry confirms the diagnosis, I'd localise the tumour with CT or MRI adrenals. If extra-adrenal disease is suspected, or in SDH-mutated cases, I'd arrange 68Ga-DOTATATE PET/CT. Finally, genetic testing is recommended for all patients to identify hereditary syndromes."
Q2: Why is preoperative alpha-blockade essential, and what happens if you give beta-blockers first?
"Alpha-blockade is essential to prevent intraoperative hypertensive crisis, which can cause stroke, MI, or arrhythmias. I'd use phenoxybenzamine (10 mg BD, titrated up) or doxazosin, starting at least 2 weeks preoperatively, aiming for BP less than 130/80 and mild orthostatic hypotension. Critically, beta-blockers must NEVER be given first or alone because this blocks beta-2 mediated vasodilation, leaving alpha-mediated vasoconstriction unopposed, precipitating severe hypertension. Beta-blockers are only added after adequate alpha-blockade if the patient has persistent tachycardia or arrhythmias."
Q3: What genetic syndromes are associated with phaeochromocytoma?
"Up to 40% of phaeochromocytomas are hereditary. The major syndromes are MEN2A and 2B—caused by RET mutations—where 50% develop bilateral phaeochromocytomas and are also at risk of medullary thyroid cancer. VHL disease causes phaeochromocytomas in 10-20%, along with cerebellar haemangioblastomas and renal cell carcinoma. NF1 has a 1-5% phaeochromocytoma risk. Finally, SDH mutations—particularly SDHB—are associated with paragangliomas and have a 30-70% malignancy risk. Genetic testing is now recommended for all patients to enable cascade screening of relatives."
Q4: How is malignant phaeochromocytoma defined and managed?
"Malignancy in phaeochromocytoma is defined by the presence of metastases in non-chromaffin tissue, such as lymph nodes, liver, lung, or bone—there are no reliable histological features to predict malignancy. Overall, 10-17% are malignant, but this rises to 30-70% in SDHB mutations. Management includes surgical debulking if resectable, 131I-MIBG radiotherapy for MIBG-avid metastases, and peptide receptor radionuclide therapy with 177Lu-DOTATATE for somatostatin receptor-positive tumours. Systemic therapy with sunitinib has shown benefit in the recent FIRSTMAPPP trial. Prognosis is variable, with 5-year survival of 40-60%."
Q5: What are the perioperative complications and how do you manage them?
"The main intraoperative complication is hypertensive crisis during tumour manipulation, managed with IV phentolamine boluses or sodium nitroprusside infusion. Arrhythmias can occur, treated with lidocaine or beta-blockers. Postoperatively, hypotension is very common due to loss of catecholamine drive and residual alpha-blockade—I'd manage this with large-volume IV fluids and vasopressors if needed. Hypoglycaemia can occur as catecholamine-mediated glycogenolysis ceases, so regular glucose monitoring and IV dextrose may be required. If bilateral adrenalectomy, acute adrenal insufficiency must be prevented with IV hydrocortisone."
Common Mistakes to Avoid
❌ Beta-blockade before or without alpha-blockade → hypertensive crisis ❌ Forgetting genetic testing → miss opportunity for cascade screening ❌ Not screening adrenal incidentalomas biochemically → miss diagnosis ❌ Assuming "rule of 10s" still applies to hereditary cases → underestimate genetic risk (now 40%, not 10%) ❌ Relying on catecholamines instead of metanephrines → lower sensitivity due to episodic secretion ❌ Assuming histology can predict malignancy → only metastases define malignancy
High-Yield Exam Pearls
✅ Plasma free metanephrines = gold standard (sensitivity 96-99%) ✅ Alpha-block FIRST, then beta-block → prevent unopposed alpha-stimulation ✅ Genetic testing for ALL → 40% hereditary ✅ SDHB mutation → highest malignancy risk (30-70%) ✅ MEN2 → bilateral phaeochromocytoma in 50%; screen for medullary thyroid cancer ✅ 68Ga-DOTATATE PET → best for paragangliomas, SDH-mutated, metastatic disease ✅ Lifelong follow-up → annual metanephrines; recurrence 5-15% ✅ "Light bulb sign" on T2 MRI → classic but not universal ✅ Postoperative hypotension → most common complication; manage with fluids ✅ Catecholamine cardiomyopathy → reversible with treatment [18]
Last Updated: 2026-01-07
Frequently asked questions
Quick clarifications for common clinical and exam-facing questions.
When should I seek emergency care for phaeochromocytoma and paraganglioma?
Seek immediate emergency care if you experience any of the following warning signs: Hypertensive crisis or paroxysmal severe hypertension, Classical triad: headache, sweating, palpitations, Resistant hypertension (less than 3 antihypertensives), Hypertension in young patient (less than 40 years), Adrenal incidentaloma with symptoms, Family history of MEN2, VHL, or SDH mutations.
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Adrenal Anatomy and Physiology
- Catecholamine Biosynthesis
Differentials
Competing diagnoses and look-alikes to compare.
- Essential Hypertension
- Anxiety Disorders and Panic Attacks
- Thyrotoxicosis
Consequences
Complications and downstream problems to keep in mind.
- Hypertensive Crisis
- Cardiomyopathy (Catecholamine-Induced)