Paeds · fetal-neonatal-and-perinatal
Hearing loss in high-risk neonates
Also known as Neonatal hearing loss · Congenital hearing impairment · Newborn hearing screening · Auditory neuropathy spectrum disorder in the NICU · JCIH risk indicators
Fellowship guide to hearing loss in high-risk neonates — the JCIH 1-3-6 screening pathway, why the NICU infant is screened with AABR rather than OAE, the risk indicators that demand ongoing surveillance, and the early-intervention ladder that protects language development when permanent loss is confirmed.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Related topics
Overview & Definition
A mother holds her two-week-old baby in the neonatal unit corridor while a screen prints a result neither parent can read. The nurse explains that the baby will need a repeat test, and a quiet panic sets in — is the child deaf? The paediatrician's job at that moment is to translate a screening result into a plan, and the skill lies in knowing which babies need watching, which need confirming, and which need not worry at all. Hearing loss in high-risk neonates is the prototype of a condition where the screen is only the beginning of the story. [2]
Permanent childhood hearing loss affects roughly one to three per thousand live births in the well population, but the rate climbs steeply in the neonatal intensive care unit, where as many as two to four per cent of graduates carry permanent loss. The high-risk neonate is the infant whose exposure — prematurity, ototoxic medication, severe jaundice, asphyxia, congenital infection, or a syndromic family — places them in a category where universal screening alone is insufficient and ongoing surveillance is mandatory. [6]
The definition examiners expect separates the type of loss from its consequence. Permanent hearing loss is a sensory deficit of the cochlea or auditory pathway that is present at birth or acquired in the perinatal period and that, if undetected, impairs the acquisition of spoken language during the critical first years of life. The aim of early detection is not the diagnosis itself but the protection of language, because the brain's auditory cortex is most plastic in the first six months, and identification after that window measurably degrades outcomes. [1]
Epidemiology & Risk Factors
The gradient is the single most useful number to hold in your head. Permanent childhood hearing loss affects one to three per thousand well newborns but rises to two to four per cent of NICU graduates, so an infant who has spent time on the unit is ten to forty times more likely to carry loss than a baby in the well-baby nursery. This is why the high-risk population, though a small fraction of all births, accounts for a disproportionate share of confirmed permanent loss in childhood. [6]
The risk indicators are codified by the Joint Committee on Infant Hearing and are worth memorising as the framework that drives surveillance. The most important for the neonatologist are a NICU stay of more than five days — which captures most extremely preterm and ventilated infants — and any use of extracorporeal membrane oxygenation, because ECMO is associated with a strikingly high rate of both sensorineural loss and auditory neuropathy. [7]
The remaining indicators cluster into four groups. Care-related exposures include ototoxic drugs — aminoglycosides such as gentamicin and loop diuretics such as furosemide given in multiple courses — and severe hyperbilirubinaemia at or near exchange transfusion levels. Perinatal insults include hypoxic-ischaemic encephalopathy and assisted ventilation beyond five days. Congenital infection, above all cytomegalovirus, is the leading non-genetic cause, and the family-history and physical groups include a parent or sibling with permanent childhood loss, craniofacial anomalies involving the ear or temporal bone, and syndromes known to include hearing loss. [7] [8]
The natural history is the part that catches candidates. Roughly half of permanent loss is genetic, about a quarter is environmental with congenital CMV dominant among these, and a meaningful fraction is progressive or late-onset. Congenital CMV in particular can produce hearing loss that is absent at birth, emerges in the first year, and worsens through early childhood, which is precisely why a single pass on the newborn screen cannot be the end of surveillance for a high-risk infant. [8]
Pathophysiology
To understand why the NICU population is screened differently, follow sound along the auditory pathway and note where each neonatal insult strikes. Sound enters the outer and middle ear, is transduced into electrical signals by the inner hair cells of the cochlea, travels along the auditory nerve, and is relayed through the brainstem nuclei that generate the auditory brainstem response. The screening tools probe different parts of this chain, and the choice between them is dictated by which part the neonatal insult damages. [5]
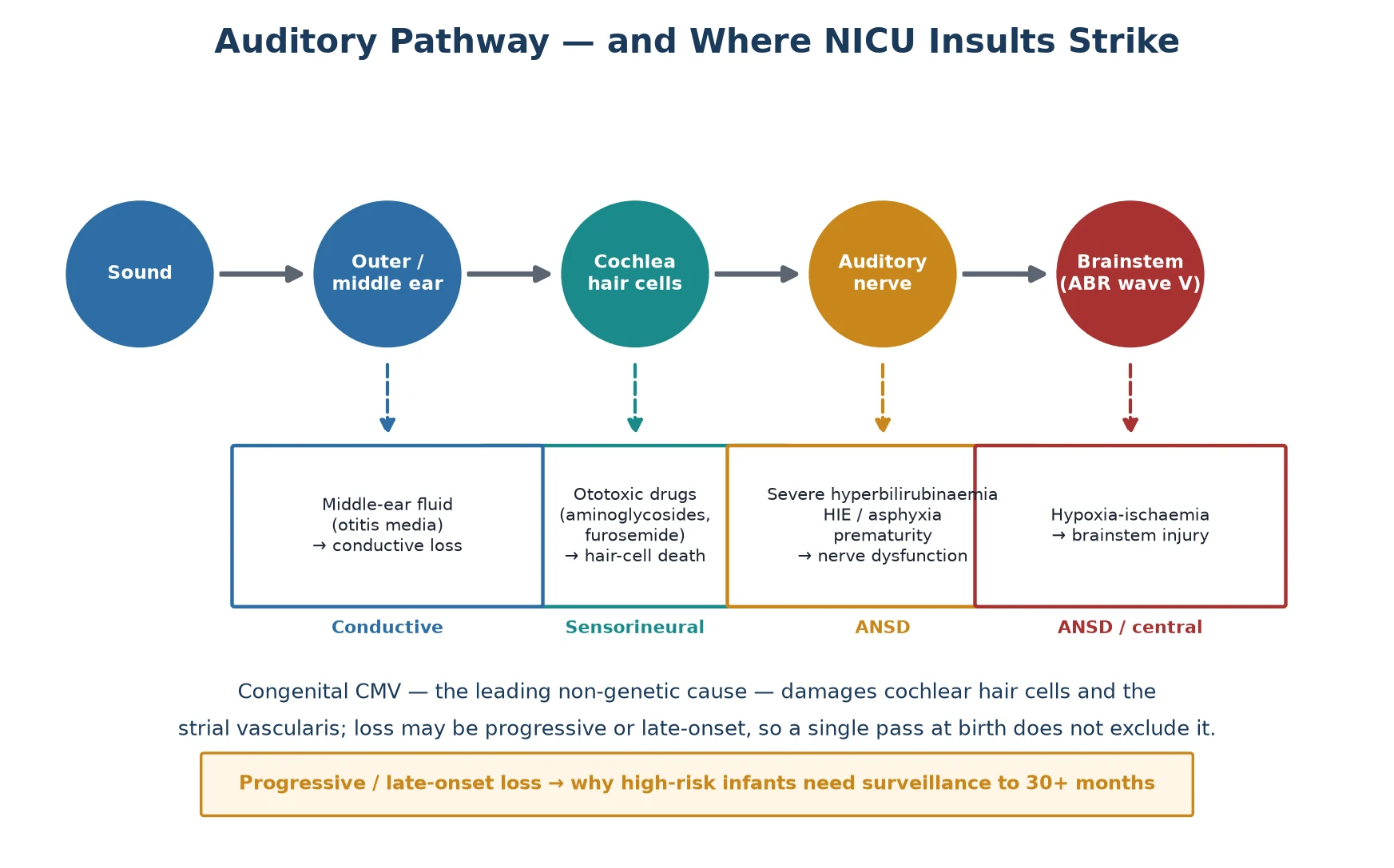
Ototoxic injury is the classical sensorineural mechanism. Aminoglycosides accumulate in and destroy the outer hair cells of the cochlea, and loop diuretics potentiate this damage by altering the endocochlear potential; the combination of gentamicin and furosemide given repeatedly to a very preterm infant is the prototypical scenario. Because the outer hair cells generate otoacoustic emissions, their destruction flattens the OAE response — which is why OAE can detect this kind of loss. [5]
Auditory neuropathy spectrum disorder is the mechanism that defines the high-risk population. Here the cochlea, including the outer hair cells, is structurally intact, so OAE is normal, but the inner hair cells, the synapse, or the auditory nerve fail to transmit a coherent signal to the brainstem. The result is a child who has a present OAE but an absent or profoundly abnormal ABR — a dissociation that OAE-only screening will entirely miss. ANSD is concentrated in the NICU precisely because its causes are the unit's signature insults: severe hyperbilirubinaemia, perinatal asphyxia and hypoxic-ischaemic injury, extreme prematurity, and congenital infection. [10] [11]
Congenital CMV operates through a third mechanism that is both sensorineural and progressive. The virus infects the cochlear hair cells and the stria vascularis, producing a loss that may be present at birth, emerge later, or fluctuate and worsen over years. This progressive biology is the reason a single negative screen at birth never closes the file for a CMV-exposed infant, and it is the reason antiviral treatment, when given early, can change the trajectory of the loss. [8]
Classification
Hearing loss is classified by the site of the lesion, and the classification is not academic — it determines which screening test will detect it and which intervention will work. Conductive loss is a mechanical problem of the outer or middle ear, seen in the newborn as aural atresia or, far more commonly in the follow-up clinic, persistent otitis media with effusion. It is generally amenable to medical or surgical correction and is the least concerning type, though it still impairs language if it persists. [6]
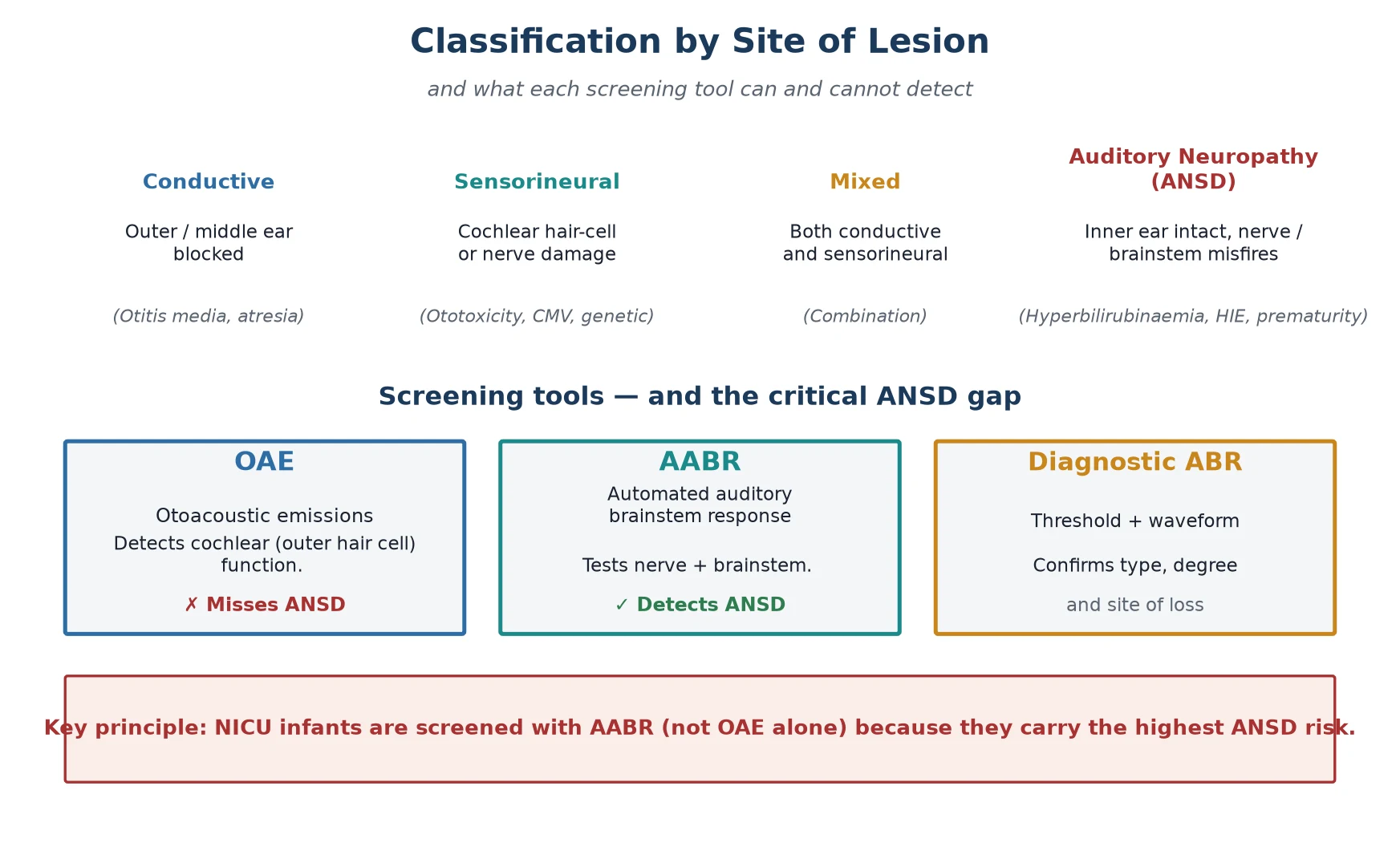
Sensorineural loss is damage to the cochlea or auditory nerve — the territory of ototoxicity, congenital CMV, and genetic causes such as connexin 26 (GJB2) mutations. Mixed loss combines both. The fourth type, auditory neuropathy spectrum disorder, is the one that demands attention: the cochlea is intact, the outer hair cells function, but the nerve or synapse misfires. ANSD accounts for a substantial minority of permanent loss in children and is heavily over-represented in the NICU, which is the central reason the high-risk infant is screened with a test that OAE cannot replace. [10]
The practical classification that changes practice is the OAE-versus-AABR distinction. Otoacoustic emissions test only the cochlear outer hair cells and will return a normal result in an infant with ANSD who has a profound functional hearing deficit. Automated auditory brainstem response tests the entire pathway from sound to brainstem, so it captures ANSD and is therefore the mandated screen for the NICU infant. Knowing which test was used — and why — is the classification question examiners ask most often. [5] [7]
Clinical Presentation
In the newborn period there is no clinical presentation to detect at the bedside, and this is the whole justification for screening. A baby with profound bilateral hearing loss looks and behaves like any other newborn: they startle to loud sounds poorly or not at all, but no parent and no clinician can reliably distinguish this from normal variability in the nursery. The absence of a reliable clinical sign is exactly why universal screening exists, and why relying on parental concern or bedside behavioural testing is now considered substandard. [2]
The presentation that does reach the paediatrician is the screening result itself — a refer on the AABR or OAE, or a missed screen at discharge. The clinical skill at this point is not in detecting hearing loss but in triaging the result: distinguishing the transient failure from the probable loss, and ensuring the infant reaches the diagnostic clinic within the window that protects language. A refer on one ear, a refer in a well baby who was unsettled during the test, or a screen done with debris in the ear canal are common and often resolve on retest. [5]
The presentation later in infancy, when a loss has been missed or is progressive, is developmental rather than auditory. The parents describe a baby who does not turn to sound, who is not babbling by nine to twelve months, or whose language is stagnating. Congenital CMV classically presents this way — a child whose newborn screen passed but who loses hearing in the first year — and the lesson is that any developmental concern about language in a high-risk infant warrants an audiological reassessment, regardless of a prior normal screen. [8]
Differential Diagnosis
The differential falls into two frames: why a screen failed, and why a child has hearing loss. A referred screen is most often explained by transient outer or middle-ear conditions that are not permanent loss. Residual vernix or amniotic fluid in the ear canal in the first day, ambient noise and infant movement during the test, and middle-ear effusion from recent respiratory illness can all produce a refer that resolves on repeat testing. These transient causes account for the majority of refers and are the reason a single refer is repeated before it becomes a referral. [5]
When loss is confirmed, the differential of cause guides prognosis, counselling, and sometimes treatment. Genetic causes predominate overall, with connexin 26 (GJB2) the single most common, followed by syndromic causes such as Pendred, Usher, and Waardenburg. Among non-genetic causes congenital CMV is the leading single agent, followed by the NICU exposures — ototoxicity, hyperbilirubinaemia, asphyxia — that produce sensorineural loss and auditory neuropathy. Identifying the cause matters because it predicts progression, recurrence risk, and the possibility of antiviral treatment. [8]
The dissociation pattern is worth naming explicitly because it points straight to the diagnosis. A child with absent ABR but present OAE has auditory neuropathy spectrum disorder until proven otherwise, and the differential within ANSD is the hyperbilirubinaemia-asphyxia-prematurity triad of the NICU, supplemented by genetic OTOF mutations and congenital infection. Recognising this pattern is what prevents the dangerous error of declaring an ANSD infant to have normal hearing on the basis of a normal OAE. [10]
Clinical & Bedside Assessment
Assessment begins with reading the screening record, because the type of test and the conditions under which it was done change everything. Confirm whether the screen was AABR or OAE, whether one or both ears referred, and whether the test was done in optimal conditions. An OAE refer in a well baby done within the first twenty-four hours, in a noisy nursery, carries a very different meaning from a bilateral AABR refer in a long-stay NICU infant who has had multiple ototoxic courses. [5]
The bedside examination, while it cannot confirm loss, contributes two things: it identifies the visible risk indicators and it excludes a transient cause. Inspect the pinnae and the ear canal for craniofacial anomaly, atresia, pits, and tags, any of which raises the probability of associated middle and inner ear malformation. Examine for dysmorphism that suggests a syndrome, palpate for a goitre or thyroid mass in Pendred, and assess the overall neurological state, because hypotonia or developmental delay may signal a syndromic or metabolic cause. [7]
Pneumatic otoscopy, where feasible, screens for middle-ear effusion as a reversible cause of a conductive refer, though it is difficult in the newborn and usually deferred to the audiologist. The remainder of the assessment is a structured review against the JCIH risk indicators: document the NICU length of stay and the ototoxic drugs given, the peak bilirubin and whether it reached exchange levels, any HIE grade, the congenital infection screen results, and a three-generation family history of hearing loss. This risk inventory is what determines whether the infant needs one screen or years of surveillance. [7]
Investigations
The screening investigation is the AABR or OAE, performed ideally before discharge and no later than one month of age. The AABR delivers a click stimulus and measures the automated pass or refer of the brainstem response, and it is the mandated modality for the NICU infant because it detects ANSD. The OAE, measuring cochlear outer hair cell function, is acceptable for the well baby but inadequate alone for the high-risk infant. A refer on the initial screen is repeated once, and a second refer triggers referral for diagnostic assessment. [7]
The diagnostic investigation is the full diagnostic auditory brainstem response, performed under sedation or in natural sleep, by a paediatric audiologist by three months of age. Unlike the screen, the diagnostic ABR is threshold-seeking and frequency-specific, allowing the audiologist to define the degree, configuration, and type of loss in each ear, and to characterise the ABR waveform for evidence of auditory neuropathy. It is paired with behavioural observation audiology as the child matures, and together they constitute the diagnostic confirmation that mandates intervention. [5]
The aetiological work-up runs in parallel and is tailored to the likely cause. A genetic panel including GJB2 (connexin 26) and a referral to clinical genetics are first-line, because genetic causes predominate. A urine CMV PCR must be sent before three weeks of age to confirm congenital infection, because after that point distinguishing congenital from acquired CMV is impossible — this time limit is a favourite exam point. Cranial imaging with MRI of the temporal bone and brain identifies inner-ear malformations and brainstem injury, and an ophthalmology review screens for the retinal changes of syndromic causes such as Usher syndrome. [8]
Management — Resuscitation
There is no acute resuscitation in the traditional sense, because hearing loss does not threaten the airway or circulation in the newborn period. The "resuscitation" here is the protection of the diagnostic timeline: ensuring that a referred infant is not lost to follow-up, that the repeat screen happens within weeks, and that the diagnostic clinic appointment is booked before the child ages out of the window that protects language. Loss to follow-up after a refer is the single most common and most damaging failure of the whole pathway, and tracking every refer to resolution is the first management duty. [2]
When a child is identified with congenital CMV and hearing loss or other end-organ disease, the time-critical intervention is antiviral therapy. Valganciclovir, at 16 mg per kilogram twice daily for up to six months, improves audiologic and developmental outcomes in symptomatic congenital CMV disease, and it must be started in the window where it can still modify the loss. The decision to treat is made with infectious diseases input, and the early urine PCR that enabled it is the investigation that made the treatment possible. [9]
For the infant with a confirmed loss, the immediate step is referral to the early-intervention and audiology team and the fitting of amplification within the six-month window. Hearing aids are the first intervention for permanent loss of any degree that impairs access to speech, and the earlier they are fitted, the better the language outcome — so the "resuscitation" of language begins the day the diagnosis is confirmed, not when the family has finished adjusting to it. [1]
Management — Definitive & Stepwise
Definitive management is a ladder whose rungs are calibrated to the degree and type of loss and to the developmental clock. The foundation is early and appropriate amplification: hearing aids fitted by six months for permanent bilateral loss, programmed to each child's audiogram and reviewed as the child grows. The evidence for early fitting is the core of the field — children identified and amplified within the first six months acquire language at rates indistinguishable from hearing peers, whereas those identified later fall progressively behind. [1]
For severe-to-profound sensorineural loss where hearing aids provide insufficient benefit, cochlear implantation is the definitive intervention, typically evaluated around twelve months of age for bilateral profound loss. The implant bypasses the damaged cochlea and stimulates the auditory nerve directly, and outcomes are best when implantation occurs within the sensitive period for language development. For auditory neuropathy spectrum disorder, the pathway is more nuanced: hearing aids may help, but the child is monitored closely, and those who do not develop usable hearing with amplification proceed to cochlear implantation, which can be highly effective when the nerve itself is intact. [10]
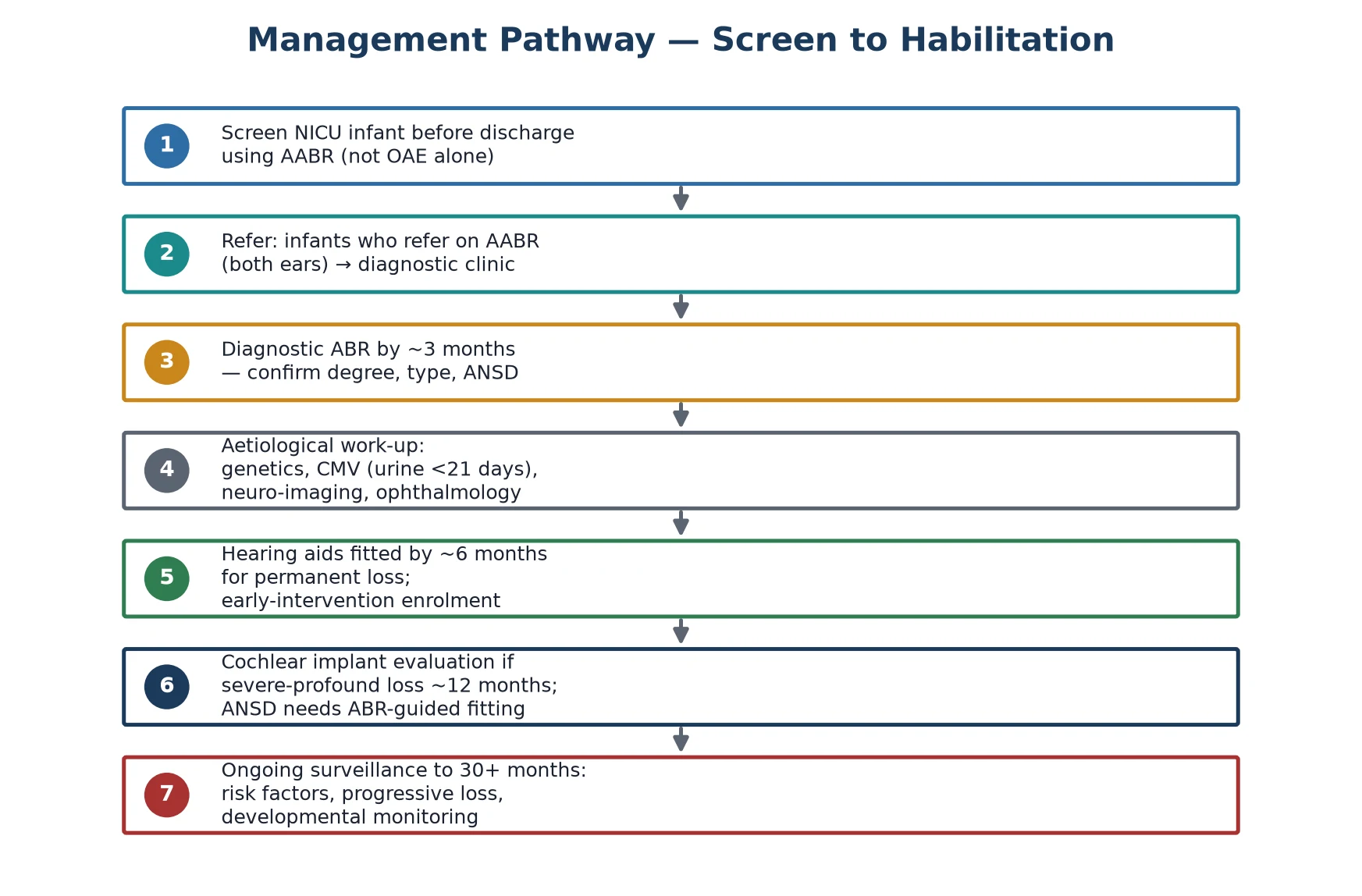
The 1-3-6 early-intervention ladder
Screen every NICU infant with AABR before discharge (well babies by 1 month)
Repeat a refer once; a second refer triggers diagnostic referral
Diagnostic frequency-specific ABR by 3 months — confirm type, degree, ANSD
Parallel aetiological work-up: genetics, urine CMV PCR before 21 days, MRI, ophthalmology
Fit hearing aids and enrol in early intervention by 6 months for permanent loss
Evaluate for cochlear implantation around 12 months if severe-profound loss
Maintain JCIH risk-indicator surveillance to at least 30 months
Beyond the device, the child and family enter an early-intervention programme that is the true engine of outcome. This includes auditory-verbal therapy or sign language support according to the family's chosen communication approach, developmental monitoring, and educational planning. The family-centred model is explicit in every guideline: the choice of communication modality belongs to the family, and the clinician's role is to inform the choice and support it, not to prescribe it. [3]
Specific Subtypes & Scenarios
Auditory neuropathy spectrum disorder is the scenario that most distinguishes the high-risk neonate, and it deserves a dedicated mental model. The infant — classically a NICU graduate who had severe jaundice or asphyxia — has a present OAE but an absent or grossly abnormal ABR, and may initially appear to hear some sounds while failing to develop speech. Management is expectant at first, with amplification trialled and hearing monitored, because some ANSD infants derive useful benefit from hearing aids; those who do not are candidates for cochlear implantation, which works because the spiral ganglion and auditory nerve are usually intact even when the synapse is not. [10] [11]
Congenital CMV is the scenario with the most actionable time window. An infant with symptomatic congenital CMV — thrombocytopenia, petechiae, hepatitis, or already-diagnosed hearing loss — should be evaluated promptly for valganciclovir therapy, because treatment within the first month of life improves both hearing and developmental outcomes. The trap is the infant whose CMV is silent at birth and whose loss appears later, which is why any infant with confirmed congenital CMV, symptomatic or not, enters audiological surveillance regardless of the newborn screen result. [9] [8]
The late-preterm and extremely preterm survivor presents a third scenario: a child who graduates the NICU with multiple risk indicators stacked together — prolonged ventilation, repeated gentamicin and furosemide courses, a patent ductus, and episodes of hypoxia — and who therefore needs not a single screen but a surveillance programme. These children are the ones in whom progressive loss emerges months or years later, and the discipline is to keep the audiology appointment on the discharge summary as a booked commitment, not an optional suggestion. [6] [7]
Complications & Pitfalls
The principal complication of undetected or late-detected hearing loss is impaired language, literacy, and social development, and this is the entire reason the screening programme exists. Children whose permanent loss is confirmed after the first six months acquire spoken language more slowly and reach lower literacy levels than those identified early, and the deficit can persist into the teenage years even when the hearing loss itself is fully habilitated. The language outcome, not the audiogram, is the measure that matters. [1] [4]
The first pitfall is screening the NICU infant with OAE alone. Because OAE tests only the cochlea, it returns normal in auditory neuropathy, and an ANSD infant with a profound functional deficit is declared to have passed. The mandated practice, codified in the guidelines, is AABR for every infant who meets the NICU criterion, and the exam point is exactly this: OAE misses ANSD, AABR catches it. [7]
The second pitfall is treating a pass as the end of the story. Congenital CMV and several genetic causes produce loss that is absent at birth and emerges later, so a pass on the newborn screen does not remove a high-risk infant from surveillance. The third is the lost-to-follow-up infant — the refer that was never repeated, the appointment that was never kept — which is the commonest real-world failure and the one that programmes are explicitly built to prevent. [2] [8]
Prognosis & Disposition
The prognosis for language is excellent when the loss is identified early and habilitated within the sensitive period. Children whose permanent bilateral loss is confirmed by three months and amplified by six months acquire receptive and expressive language at rates comparable to hearing peers, and this benefit is sustained into adolescence, where universal-screening cohorts show better language and literacy than their later-identified counterparts. The prognosis, in other words, is determined less by the severity of the loss than by the age at which it is addressed. [1] [4]
The randomised and controlled evidence underpinning universal screening is the Kennedy controlled trial and its follow-up, which demonstrated that screening reduced the age at confirmation and that early confirmation improved language outcomes through childhood. The 1-3-6 framework is the operational distillation of that evidence, and it is the standard against which every programme is measured. [2] [3]
Disposition from the neonatal unit must include the screen result, the surveillance plan, and a booked audiology appointment for any infant with a risk indicator. The high-risk infant goes home not with a cleared screen but with a surveillance schedule that may extend to thirty months or beyond, and the discharge summary is the document that prevents the loss from being rediscovered only when language fails. [7]
Special Populations
The extremely preterm and NICU graduate is the central special population, carrying the stacked risk of prolonged ventilation, ototoxic exposure, hyperbilirubinaemia, and hypoxia-ischaemia. Their hearing loss is more often auditory neuropathy, more often progressive, and more often compounded by other neurodevelopmental vulnerabilities, so they require the most intensive surveillance and the tightest follow-up of any screened group. [6]
NICU HEAR — the risk indicators to document at discharge
The infant from a socioeconomically disadvantaged or migrant-refugee background faces a different and equally important risk: loss to follow-up. These families are over-represented among the infants who refer on screening and never reach the diagnostic clinic, defeated by transport, language, and fragmented care. The intervention here is system-level — dedicated tracking, interpreter-supported appointments, and a named coordinator — because the harm of a missed diagnosis falls disproportionately on those least able to navigate the pathway. [2]
The infant with a confirmed congenital CMV infection, whether symptomatic or not, constitutes a surveillance population of its own, because CMV-related loss is the archetypal progressive loss. These children are monitored at defined intervals through the first years of life regardless of the newborn screen, and any change in hearing triggers reassessment of the antiviral question. [8]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars. The first is the controlled trial of universal newborn hearing screening by Kennedy and colleagues, published in the Lancet, which demonstrated that screening reduced the age at diagnosis and that earlier diagnosis improved language — the finding that operationalised the 1-3-6 standard. The second is the foundational work of Yoshinaga-Itano showing that early-identified children acquire language far better than later-identified ones, and the long-term follow-up by Pimperton and colleagues confirming that the benefit persists into the teenage years. The third is the programme of work on screening test performance by Norton, Vohr, and Cone-Wesson, which established that AABR detects ANSD and that OAE alone does not. [2] [1] [4] [5]
The guideline authority is the Joint Committee on Infant Hearing, whose position statements codify the risk indicators and the 1-3-6 framework. The current statement, together with its national adaptations, is the document that drives screening practice worldwide, and it is the source examiners expect you to cite for the risk-indicator list and the surveillance schedule. [7]
Regional practice differs less in principle than in execution. The 1-3-6 standard is broadly shared across Australia and Aotearoa New Zealand, the United Kingdom's NHS Newborn Hearing Screening Programme, the United States Early Hearing Detection and Intervention programmes, and the Canadian provincial equivalents. The differences that matter are in coverage, in loss-to-follow-up rates, and in access to habilitation services for rural and Indigenous families, where the principles of the guideline are often frustrated by the realities of geography and equity. [2]
Exam Pearls
The numbers to know cold are the 1-3-6 months of the JCIH pathway and the 16 mg per kilogram twice-daily dose of valganciclovir for symptomatic congenital CMV. The screening distinction is the single most tested point: OAE tests the cochlea and misses ANSD, AABR tests the whole pathway and catches it, and that is why the NICU infant is screened with AABR. [7] [9]
Hold the dissociation pattern firmly — present OAE with absent ABR is auditory neuropathy — and remember that congenital CMV demands a urine PCR before three weeks of life, after which the congenital diagnosis can no longer be made and the chance to treat a progressive loss is forfeited. The risk-indicator list, the surveillance schedule, and the reason for ongoing follow-up after a pass are the communication-circuit and short-case points that separate a pass from a distinction. [8] [10]
Finally, the overarching message is the one to carry into the viva: hearing screening exists to protect language, and the high-risk neonate is the infant for whom a single screen is never enough. The 1-3-6 framework, the AABR-not-OAE rule, the CMV time window, and the surveillance of the risk indicator are the four ideas that, named cleanly, show the examiner you understand both the screening test and the child it is meant to protect. [1] [2]
References
- [1]Yoshinaga-Itano C, Sedey AL, Coulter DK, Mehl AL Language of early- and later-identified children with hearing loss. Pediatrics, 1998.PMID 9794949
- [2]Kennedy C, McCann D, Campbell MJ, Kimm L, Thornton R Universal newborn screening for permanent childhood hearing impairment: an 8-year follow-up of a controlled trial. Lancet, 2005.PMID 16112302
- [3]Pimperton H, Kennedy CR The impact of early identification of permanent childhood hearing impairment on speech and language outcomes. Arch Dis Child, 2012.PMID 22550319
- [4]Pimperton H, Kreppner J, Mahon M, Stevenson J, Terlektsi E, Worsfold S, Yuen HM, Kennedy CR Language Outcomes in Deaf or Hard of Hearing Teenagers Who Are Spoken Language Users: Effects of Universal Newborn Hearing Screening and Early Confirmation. Ear Hear, 2017.PMID 28399063
- [5]Norton SJ, Gorga MP, Widen JE, Folsom RC, Sininger Y, Cone-Wesson B, Vohr BR, Mascher K, Fletcher K Identification of neonatal hearing impairment: evaluation of transient evoked otoacoustic emission, distortion product otoacoustic emission, and auditory brain stem response test performance. Ear Hear, 2000.PMID 11059707
- [6]Vohr BR, Widen JE, Cone-Wesson B, Sininger YS, Gorga MP, Folsom RC, Norton SJ Identification of neonatal hearing impairment: characteristics of infants in the neonatal intensive care unit and well-baby nursery. Ear Hear, 2000.PMID 11059699
- [7]American Academy of Pediatrics, Joint Committee on Infant Hearing Year 2007 position statement: Principles and guidelines for early hearing detection and intervention programs. Pediatrics, 2007.PMID 17908777
- [8]Goderis J, De Leenheer E, Smets K, Van Hoecke H, Keymeulen A, Dhooge I Hearing loss and congenital CMV infection: a systematic review. Pediatrics, 2014.PMID 25349318
- [9]Kimberlin DW, Jester PM, Sanchez PJ, Ahmed A, Arav-Boger R, Michaels MG, Ashouri N, Englund JA, Estrada B, Jacobs RF, Romero JR, Sood SK, Whitworth MS, tamburu AT, Wang J, Fowler K, et al Valganciclovir for symptomatic congenital cytomegalovirus disease. N Engl J Med, 2015.PMID 25738669
- [10]Morlet T, Parkes W, Pritchett C, Venskytis E, DeVore B, O'Reilly RC A 15-Year Review of 260 Children With Auditory Neuropathy Spectrum Disorder: I. Demographic and Diagnostic Characteristics. Ear Hear, 2023.PMID 37036288
- [11]Chen W, Huang S, Huang Y, Duan B, Xu Z, Wang Y Short-term outcomes of infants with hyperbilirubinemia-associated auditory neuropathy spectrum disorder in neonatal intensive care unit. Int J Pediatr Otorhinolaryngol, 2023.PMID 37172369