Paeds · haematology-oncology-and-transfusion
Von Willebrand disease
Also known as Von Willebrand disease · VWD · Von Willebrand factor deficiency · Vascular haemophilia
Fellowship guide to von Willebrand disease, the most common inherited bleeding disorder, caused by a quantitative or qualitative defect of von Willebrand factor. Covers the Sadler 1994 classification into type 1 (partial deficiency, about 75 to 80 percent), the type 2 qualitative subtypes (2A loss of high-molecular-weight multimers, 2B gain-of-function with thrombocytopenia, 2M normal multimers but reduced function, 2N reduced factor VIII binding), and type 3 (virtual absence, severe, autosomal recessive, about 1 in a million); the two functions of von Willebrand factor as platelet adhesion via glycoprotein Ib and as the carrier and stabiliser of factor VIII; the diagnostic panel of von Willebrand factor antigen, ristocetin cofactor activity, factor VIII, ristocetin-induced platelet aggregation and multimer analysis; the desmopressin trial at 0.3 micrograms per kg intravenously over 30 minutes; and VWF concentrate, tranexamic acid and the special care of the adolescent with heavy menstrual bleeding, the neonate and the pregnant carrier.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the thirteen-year-old girl sent to the paediatric clinic because her periods have been so heavy she has needed iron tablets and missed school, and you also learn she has always bruised easily and had nose-bleeds that lasted an hour. Her full blood count shows a mild microcytic anaemia, her platelet count is normal, and her coagulation screen is normal. That girl most likely has von Willebrand disease, and the heavy menstrual bleeding is its signature presentation in adolescence. The easy bruising and prolonged epistaxis are the mucocutaneous pattern that points to a primary-haemostasis defect rather than a clotting-factor deficiency. [1] [10]
Von Willebrand disease is an inherited bleeding disorder caused by a quantitative or qualitative defect of von Willebrand factor, a large multimeric glycoprotein that does two distinct jobs in haemostasis. It is the most common inherited bleeding disorder: the population prevalence of symptomatic disease is about 1 in 1000, though many cases are mild and never diagnosed, and the severe type 3 form affects about 1 in a million. Erik von Willebrand described the disease in 1926 in families on the Aland Islands, and it is now understood as a single disease with a spectrum of types rather than one uniform condition. [6] [1]
The defining principle is that VWF sits at the interface between primary and secondary haemostasis. It carries factor VIII in the circulation, protecting it from breakdown, and it mediates the adhesion of platelets to the injured vessel wall. So a shortage or malfunction of VWF can produce both the platelet-type bleeding of mucocutaneous surfaces and, in the severe forms, a low factor VIII that allows deeper joint and muscle bleeding. This dual role is the key to understanding why the disease looks the way it does and why the laboratory pattern varies so much between types. [4] [1]
Classification
Von Willebrand disease is classified by whether the VWF is reduced in amount or faulty in function, and the classification drives both the bleeding pattern and the treatment. The Sadler 1994 classification, endorsed by the International Society on Thrombosis and Haemostasis, divides the disease into type 1, type 2 (with four subtypes) and type 3. The amount of VWF tells you the type; the function and the multimer pattern tell you the subtype; and together they decide whether desmopressin will work or whether the child needs VWF concentrate. [5] [1]
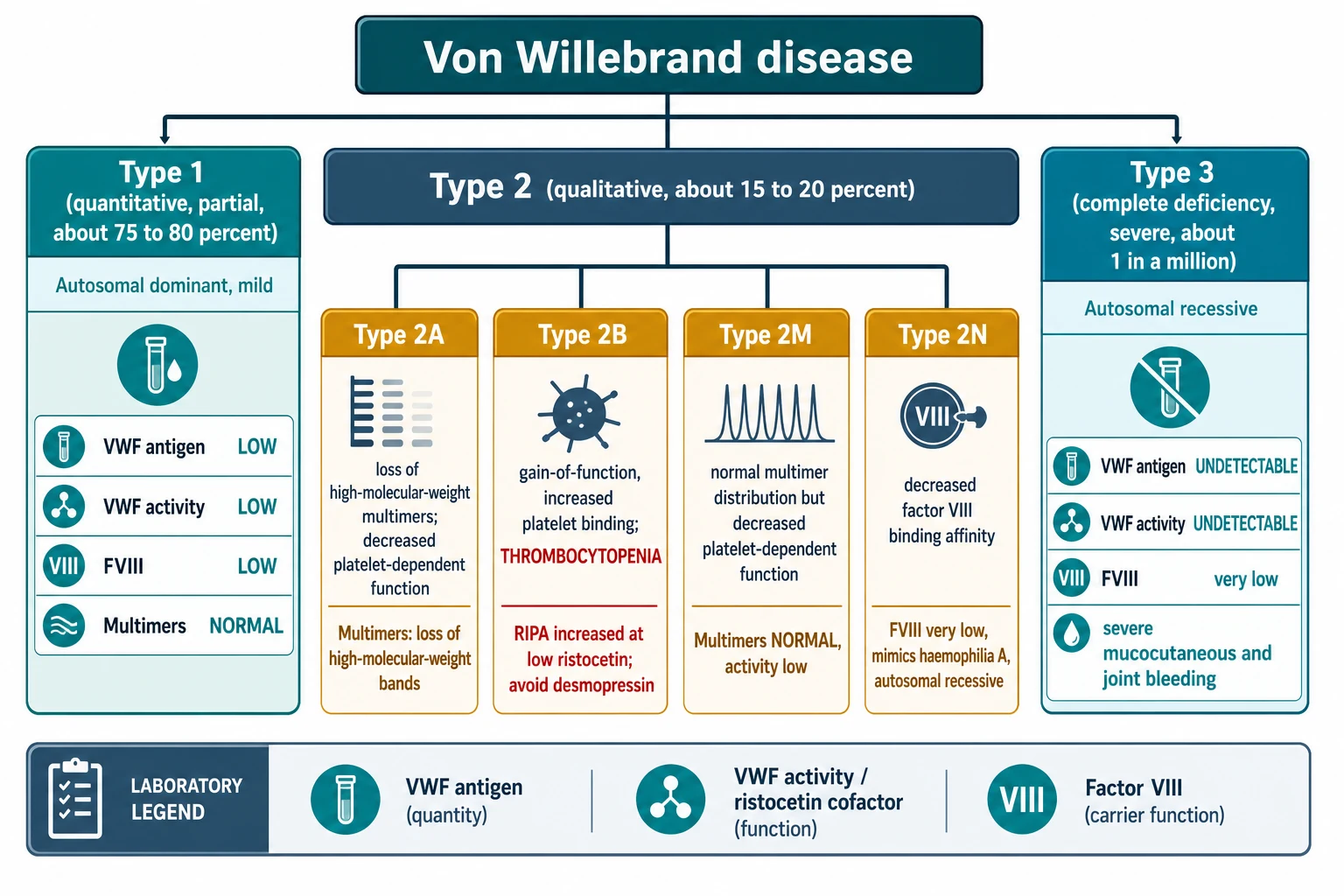
Type 1 is a partial quantitative deficiency of normally functioning VWF and accounts for about 75 to 80 percent of all cases. It is usually autosomal dominant and mild, and the VWF antigen, activity and factor VIII are all proportionally reduced while the multimer pattern stays normal. Because some functional VWF remains, most patients with type 1 respond well to desmopressin, which releases stored VWF from the endothelium. [5] [7]
Type 1
Partial deficiency, about 75 to 80 percent
- Autosomal dominant, usually mild
- VWF antigen, activity and factor VIII all low
- Multimers normal
- Usually responds to desmopressin
Type 2A
- Qualitative defect, loss of HMW multimers
- Reduced platelet-dependent function
- Activity lower than antigen
- May respond to desmopressin
Type 2B
- Gain-of-function, binds platelets too well
- Thrombocytopenia
- Increased ristocetin-induced platelet aggregation
- Desmopressin generally avoided
Type 2M
- Normal multimer distribution
- Reduced platelet-dependent function
- Activity lower than antigen
- Variable desmopressin response
Type 2N
- Reduced factor VIII binding affinity
- Factor VIII very low, mimics haemophilia A
- Autosomal recessive, both sexes
- Use VWF concentrate
Type 3
Severe, about 1 in a million
- Virtually absent VWF
- Autosomal recessive
- Severe mucocutaneous and joint bleeding
- No response to desmopressin
Type 2 is a qualitative defect and accounts for about 15 to 20 percent of cases. It subdivides into four subtypes that differ in exactly which function of VWF is broken. Type 2A has a loss of the high-molecular-weight multimers that are the most haemostatically active, so platelet-dependent function falls while the antigen may be near normal. Type 2B is a gain-of-function mutation that makes VWF bind spontaneously to circulating platelets, which both removes the high-molecular-weight multimers and causes thrombocytopenia; this is the subtype where desmopressin is generally avoided because it releases more of the abnormal VWF and drops the platelet count further. Type 2M has a normal multimer distribution but a reduced platelet-binding function, while type 2N has a reduced affinity for factor VIII so that factor VIII is lost and the picture mimics mild haemophilia A. [5] [1]
Type 3 is the rare, severe form in which VWF is virtually absent, affecting about 1 in a million people. It is autosomal recessive, the factor VIII level is very low because there is no VWF to carry it, and the bleeding is severe, including mucocutaneous bleeding and joint and muscle bleeds that resemble haemophilia. There is no stored VWF to release, so desmopressin is useless and these patients depend on VWF concentrate for both bleeds and prophylaxis. [5] [10]
Epidemiology & Risk Factors
Von Willebrand disease is the most common inherited bleeding disorder. The population study by Rodeghiero in 1987 screened Italian schoolchildren and found a prevalence of symptomatic disease of about 1 in 1000, with the great majority being mild type 1, while the severe type 3 form is much rarer at about 1 in a million. The true prevalence of low VWF in the population is higher than the symptomatic prevalence, because many people with mildly reduced VWF never bleed enough to come to medical attention. [6] [1]
The epidemiology at a glance
The single most important modifier of the VWF level is ABO blood group. People with blood group O have VWF antigen and activity about 25 to 30 percent lower than people with non-O groups, which is enough to push a borderline patient below the diagnostic threshold and to confuse the interpretation of a single result. VWF also rises with stress, exercise, inflammation and oestrogen, and it falls in hypothyroidism, so a level drawn during an acute illness or on the oral contraceptive pill may be falsely reassuring. This variability is why a single normal VWF level never excludes the disease when the bleeding phenotype fits, and repeat testing is part of the diagnostic standard. [2] [7]
The inheritance depends on the type. Types 1, 2A, 2B and 2M are usually autosomal dominant, so an affected parent often has a bleeding history and each child has a 50 percent chance of inheriting it. Type 3 and type 2N are autosomal recessive, requiring two pathogenic variants, and the parents are usually unaffected carriers. The VWF gene sits on chromosome 12, and genetic testing is increasingly used to confirm type 2 subtypes and type 3 and to support family counselling. [5] [3]
Pathophysiology
To understand von Willebrand disease, hold onto the two jobs that VWF does, because each type of the disease breaks one or both of them. VWF is a huge multimeric glycoprotein synthesised by endothelial cells, which store it in their Weibel-Palade bodies, and by megakaryocytes, which store it in platelet alpha granules. The molecule is built into ever-larger multimers, and the largest, the high-molecular-weight multimers, are the most haemostatically active because they present many binding sites at once. [1] [4]
The first job is primary haemostasis: platelet adhesion to the injured vessel wall. When the endothelium is breached, VWF unfurls from the subendothelium and binds exposed collagen through its A3 domain. Flowing blood then sweeps platelets past, and VWF catches them through their glycoprotein Ib-IX-V receptor using its A1 domain, making the platelets roll, slow and arrest on the surface so they can form the initial platelet plug. This is the step that fails when VWF is low or faulty, and its failure produces the mucocutaneous bleeding of easy bruising, epistaxis, gum bleeding and heavy menstrual bleeding. [4] [1]
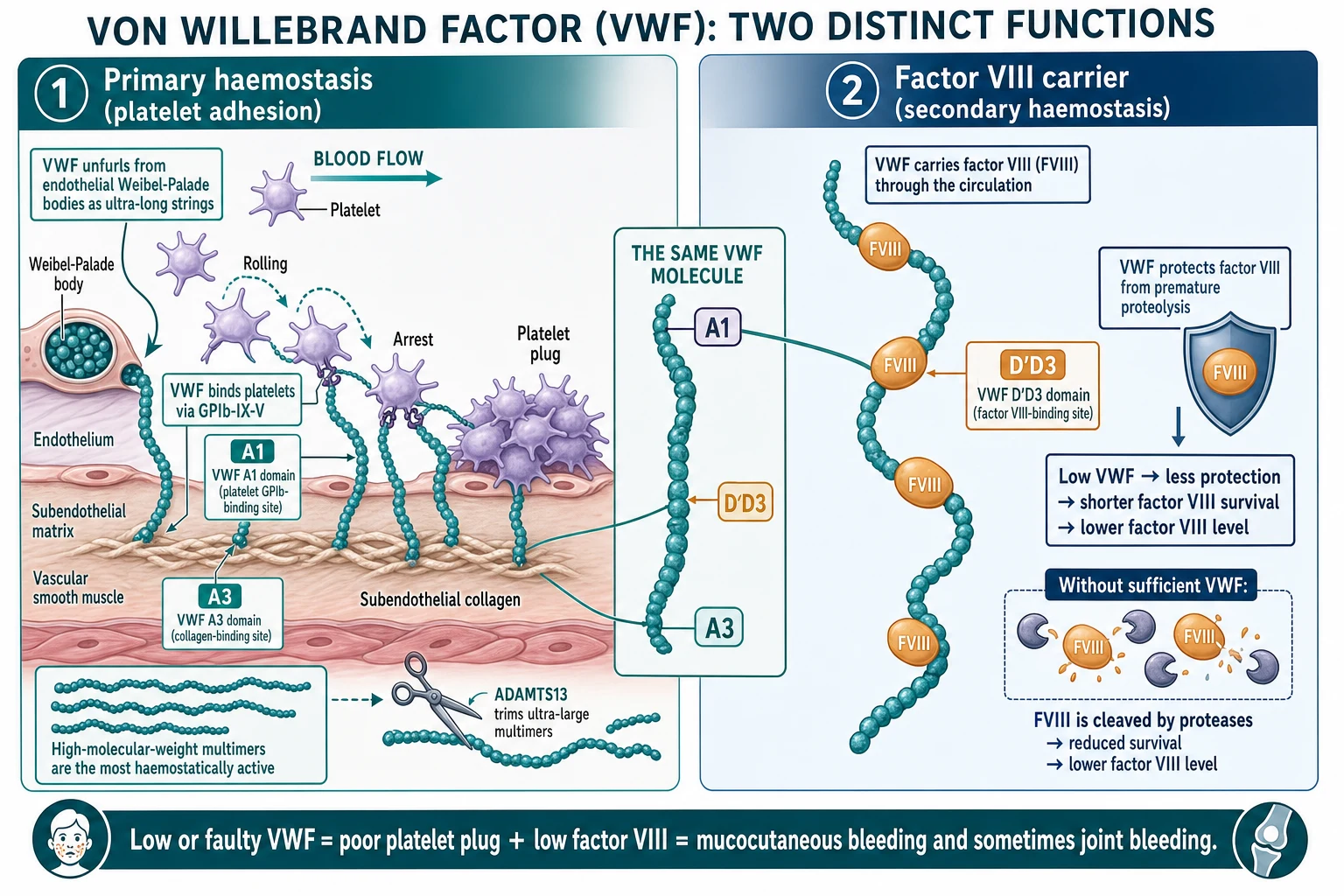
The second job is secondary haemostasis: carrying and stabilising factor VIII. VWF is the carrier protein for factor VIII in the circulation, binding it through its D prime D3 domain and protecting it from premature breakdown by proteases. This is the same relationship that makes von Willebrand disease the single most important mimic of mild haemophilia A. When VWF is low, factor VIII is cleared faster and its level falls, and in the severe type 3 form the factor VIII can drop low enough to cause joint and muscle bleeds indistinguishable from haemophilia. [4] [2]
The multimer size is actively regulated. The metalloprotease ADAMTS13 cleaves the ultra-large multimers as they enter the circulation, keeping the multimer distribution within a normal range. In type 2A the disease produces multimers that are unusually susceptible to cleavage or that fail to assemble, so the high-molecular-weight bands are lost; in type 2B the gain-of-function binding to platelets strips the large multimers out of the plasma and consumes platelets at the same time, producing thrombocytopenia. So the multimer pattern and the platelet count are not incidental findings but direct readouts of the molecular fault. [5] [1]
[4] [1]Clinical Presentation
The clinical presentation follows the type and the VWF level, but the common thread is mucocutaneous bleeding. The child bruises easily, has recurrent or prolonged nose-bleeds, bleeds for a long time after a dental extraction or the loss of a tooth, and bleeds from the gums. In adolescent females, heavy menstrual bleeding is the single most common reason the diagnosis is finally made. The platelet count and the prothrombin time are normal, and the activated partial thromboplastin time is normal or only mildly prolonged, which is why the condition is so easily missed on routine testing. [2] [10]
The typical type 1 presentation is mild mucocutaneous bleeding that becomes obvious at a challenge: prolonged bleeding after a tonsillectomy or dental extraction, heavy bleeding after childbirth, or menorrhagia in adolescence. Many patients never bleed spontaneously and are only picked up because a relative is diagnosed or because a preoperative screen prompts a closer look. A careful bleeding history is therefore the most sensitive screening tool, far more than any single laboratory value. [9] [7]
| Clinical picture | What it implies | Act |
|---|
The severe type 3 presentation is different and dangerous. Because VWF is virtually absent and factor VIII is very low, these children bleed from mucocutaneous surfaces and also into joints and muscles, much like severe haemophilia. They may present in infancy or early childhood with severe epistaxis, gastrointestinal bleeding, haematomas and joint bleeds, and they are at risk of life-threatening bleeding after trauma or surgery. A small number of type 3 patients develop alloantibodies against infused VWF after repeated exposure, which makes treatment much harder. [10] [11]
The neonatal presentation is usually silent, because most neonates with type 1 or type 2 have enough VWF to avoid bleeding at birth. Type 3 can present in the neonate with a cephalohaematoma, umbilical stump bleeding or prolonged mucosal bleeding, and infants born to mothers with type 2B can have transient thrombocytopenia from the transplacental passage of the abnormal VWF. Any neonate with unexplained mucocutaneous or umbilical bleeding deserves a VWF panel rather than being dismissed as a minor bleeding tendency. [1] [10]
Differential Diagnosis
Build the differential around the single abnormal pattern that von Willebrand disease produces: mucocutaneous bleeding with a normal platelet count and a normal or near-normal coagulation screen. From there, the question is whether the fault is in VWF, in platelet function, in a clotting factor, or in the vessel wall. The bleeding history and a focused panel usually separate them. [1] [3]
The most important mimic to separate is mild haemophilia A. Both von Willebrand disease and mild haemophilia A can present with a low factor VIII, but they differ in the bleeding pattern and the family history. Von Willebrand disease causes mucocutaneous bleeding, affects both sexes, and is usually autosomal dominant, while haemophilia A causes delayed deep joint and muscle bleeding, affects males, and is X-linked recessive. The decisive test is the VWF panel: it is low in von Willebrand disease and normal in haemophilia A. Always send VWF antigen and activity before labelling a child as mild haemophilia A. [4] [2]
Von Willebrand disease
- Autosomal, both sexes
- Mucocutaneous bleeding
- Low VWF antigen and activity
- Factor VIII may be low
Mild haemophilia A
- X-linked recessive, males
- Joint and muscle bleeds
- Low factor VIII
- VWF antigen and activity normal
Platelet function defect
- Autosomal, both sexes
- Mucocutaneous bleeding
- VWF panel normal
- Platelet function testing abnormal
Immune thrombocytopenia
- Acquired, both sexes
- Petechiae and mucosal bleeding
- Low platelet count
- Normal VWF panel
Type 2N VWD
- Autosomal recessive
- Factor VIII very low
- Looks like haemophilia A
- Abnormal factor VIII binding assay
Platelet function defects are the next group: they produce the same mucocutaneous bleeding with a normal count and a normal coagulation screen, and they are separated by platelet function testing, which is abnormal while the VWF panel is normal. Immune thrombocytopenia produces petechiae and mucosal bleeding but is distinguished by a low platelet count, and it behaves acutely rather than lifelong. Connective tissue disorders such as Ehlers-Danlos syndrome cause easy bruising and bleeding from tissue fragility and are suggested by joint hypermobility and skin hyperextensibility. [3] [2]
Finally, distinguish acquired von Willebrand syndrome, in which a previously well person develops a low or dysfunctional VWF because of an underlying condition. This is seen with hypothyroidism, with lymphoproliferative and myeloproliferative disorders, and characteristically with mechanical circulatory support such as a ventricular assist device, where high shear stress unravels and cleaves the high-molecular-weight multimers. Acquired disease has no lifelong bleeding history and resolves when the underlying cause is treated, and recognising it changes the management entirely. [1] [10]
Clinical & Bedside Assessment
Assessment begins with a structured bleeding history, because the pattern of bleeding is more sensitive than any single laboratory value. Use a validated bleeding assessment tool such as the ISTH bleeding assessment tool, which scores the number and severity of bleeding symptoms across mucocutaneous, surgical, dental and menstrual domains. A high bleeding score in a child with a borderline VWF level pushes the diagnosis toward von Willebrand disease, while a low score with a mildly reduced level suggests a non-diseased low VWF that does not need treatment. [8] [9]
Ask specifically about epistaxis (frequency, duration, whether it needs pressure or packing), gum and dental bleeding, bruising (site, size, whether it is raised), bleeding after surgery or dental extraction, postpartum bleeding, and heavy menstrual bleeding. For the adolescent female, quantify the menstrual loss: cycle length, number of pads or tampons, flooding, clots, and whether the bleeding interferes with school, sport or social life, because heavy menstrual bleeding severe enough to cause iron deficiency or disrupt daily life is a strong pointer to a bleeding disorder. Take a three-generation family history for easy bruising, nose-bleeds, surgical bleeding, postpartum haemorrhage and heavy periods, which trace the autosomal dominant pattern. [2] [8]
Examination is usually normal apart from the signs of bleeding and its consequences. Look for bruises, petechiae (which would point to thrombocytopenia rather than VWD), evidence of recent epistaxis or gum bleeding, and signs of iron deficiency anaemia such as pallor. Examine the joints and skin for the hypermobility and hyperextensibility of Ehlers-Danlos syndrome, and assess for the hepatosplenomegaly or lymphadenopathy that might suggest an acquired cause. In type 3 there may be signs of chronic anaemia and, after years of joint bleeds, the arthropathy that mirrors haemophilia. [3] [10]
[2] [10]Investigations
The investigation strategy has three steps: confirm that the bleeding is from a VWF defect, classify the type, and exclude the mimics. The first-line panel measures VWF antigen, VWF activity and factor VIII activity, all reported in IU per mL or as a percentage of normal. In type 1 all three are proportionally reduced; in the type 2 subtypes the activity is lower than the antigen; and in type 3 the VWF is virtually undetectable and the factor VIII is very low. [2] [1]
VWF antigen measures the quantity of VWF protein. VWF activity is most often measured as the ristocetin cofactor activity, which tests how well the VWF supports platelet agglutination in the presence of ristocetin; newer assays that measure binding to a recombinant glycoprotein Ib fragment are increasingly used because they are more reliable and less variable. The ratio of activity to antigen is a key number: a ratio below about 0.6 suggests a type 2 qualitative defect and prompts multimer analysis. Factor VIII is measured because VWF stabilises it, and a low factor VIII with mucocutaneous bleeding is von Willebrand disease until proven otherwise. [1] [2]
The VWF diagnostic panel and what each test measures
Classifying the subtype requires the second-line tests. Ristocetin-induced platelet aggregation tests the patient's platelet-rich plasma response to ristocetin: an exaggerated response at a low ristocetin concentration is the signature of type 2B, because the gain-of-function VWF agglutinates platelets too readily. Multimer analysis by gel electrophoresis shows the loss of the high-molecular-weight bands in type 2A and type 2B and a normal distribution in type 2M. The VWF factor VIII binding assay confirms type 2N by showing reduced affinity of VWF for factor VIII. The platelet count matters too, because thrombocytopenia is a clue to type 2B. [5] [2]
Desmopressin acetate
Dose
0.3 micrograms per kg intravenously over 30 minutes; Measure VWF activity and factor VIII at baseline, then at 1 hour and 4 hours; Expected rise of 2 to 4 times the baseline level; Adequate response defined as VWF activity and factor VIII reaching at least 0.50 IU per mL at peak
The diagnosis is not made on a single sample. VWF levels fluctuate with blood group, stress, inflammation, oestrogen and thyroid function, so a normal result in a patient with a convincing bleeding history must be repeated, ideally on two or three occasions and when the patient is well. A VWF antigen or activity below 0.30 IU per mL is definitive for type 1 disease, while a level between 0.30 and 0.50 IU per mL is a borderline zone that is interpreted together with the bleeding score and the family history rather than treated as a binary diagnosis. Genetic testing is increasingly used for the type 2 subtypes and type 3, and to support family counselling. [7] [3]
Management — Resuscitation
The resuscitation principle for an acute severe bleed in von Willebrand disease is to raise the VWF activity and factor VIII to a haemostatic level without delay. For a major or life-threatening bleed this means VWF concentrate, not desmopressin, because desmopressin takes time to work, releases only a limited store, and is ineffective or harmful in the severe types. Target a VWF activity and factor VIII above 50 IU per dL for most major bleeds, and above 80 to 100 IU per dL for life-threatening bleeding or major surgery. [2] [4]
Resuscitation of a severe bleed in von Willebrand disease
Recognise major bleeding: uncontrolled epistaxis, gastrointestinal or genitourinary bleeding, surgical bleeding, or a deep bleed in a type 3 patient
Give VWF concentrate to raise activity and factor VIII above 50 IU per dL (above 80 to 100 IU per dL for life-threatening bleeding)
Add tranexamic acid 15 mg per kg orally or 10 mg per kg intravenously three times daily for mucosal bleeding
Apply local measures: nasal packing for epistaxis, local haemostatic agents for dental sockets
Do NOT give desmopressin in type 2B or type 3, and contact the haemophilia or bleeding-disorder centre for ongoing dosing
Tranexamic acid is a valuable adjunct in the resuscitation of mucosal bleeding. It inhibits plasminogen activation and so stabilises clot breakdown at mucosal surfaces where fibrinolysis is active, which is why it is especially effective for epistaxis, dental bleeding, heavy menstrual bleeding and gastrointestinal bleeding. Give 15 mg per kg orally or 10 mg per kg intravenously three times daily, and combine it with local measures such as nasal packing and haemostatic agents. Avoid tranexamic acid in haematuria, because clot retention in the renal tract can cause obstruction. [4] [2]
The general measures apply alongside specific therapy: avoid intramuscular injections and aspirin or non-steroidal anti-inflammatory drugs, which worsen bleeding; give vaccines and analgesia by the subcutaneous or intravenous route; and provide analgesia and fluid balance. A child with von Willebrand disease who is bleeding should never receive a non-steroidal anti-inflammatory drug for pain, and paracetamol is the preferred analgesic. For a child with an alloantibody to VWF (rare, in type 3), recombinant activated factor VII or a bypassing approach may be needed, guided by a specialist centre. [2] [11]
Management — Definitive & Stepwise
Definitive management runs along three lines matched to the type: desmopressin for the responsive patient, VWF concentrate for the severe or non-responsive patient, and the non-replacement adjuncts of tranexamic acid and hormonal control for mucosal and menstrual bleeding. The choice is driven by the type and by whether the patient has responded to a desmopressin test, and the aim is to use the least invasive effective treatment while keeping VWF concentrate for those who genuinely need it. [4] [3]
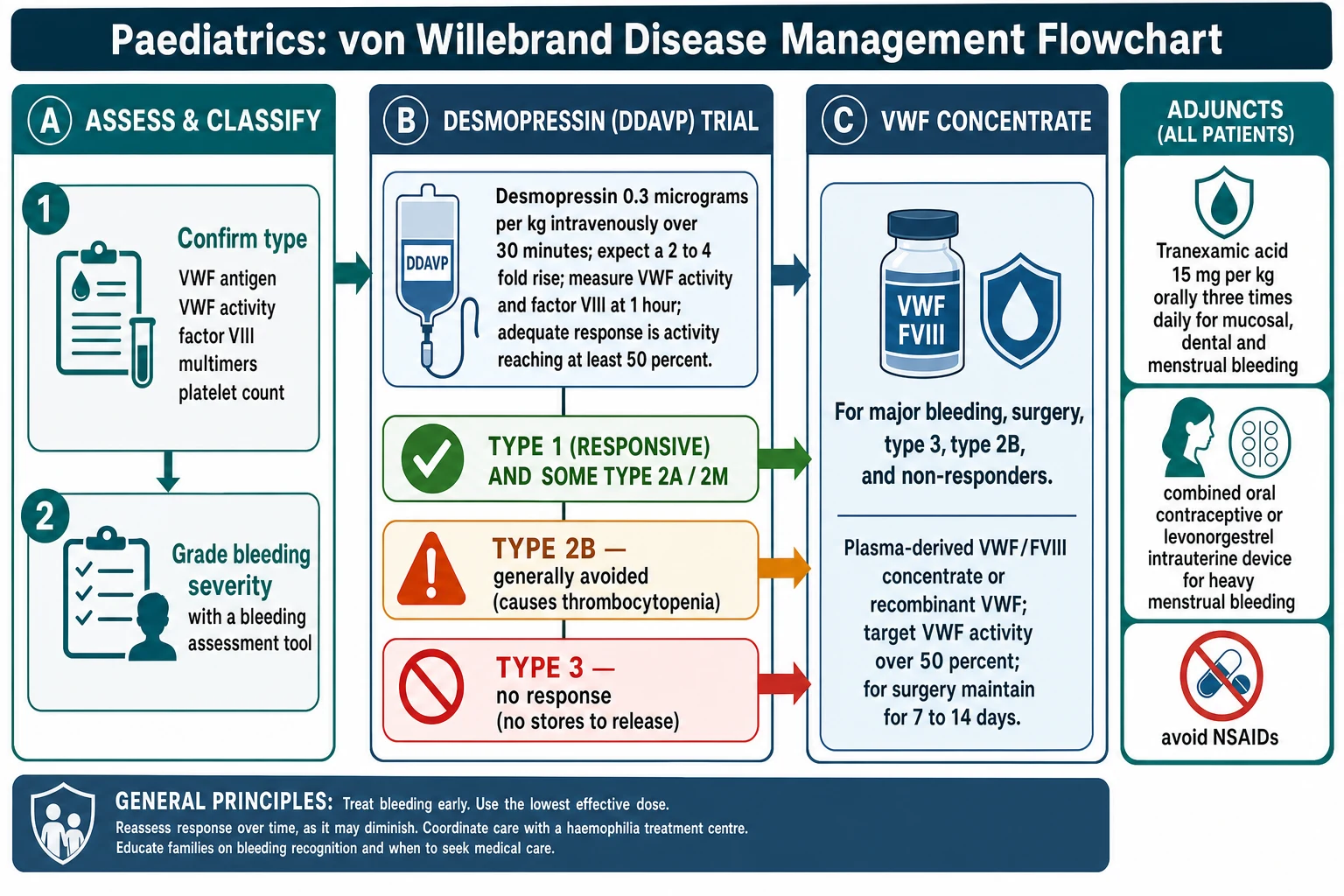
Desmopressin is first-line for the responsive patient. A dose of 0.3 micrograms per kg intravenously over 30 minutes releases stored VWF and factor VIII from endothelial Weibel-Palade bodies, typically raising both by 2 to 4 times baseline, and the effect lasts for 8 to 12 hours. An adequate response is defined as the VWF activity and factor VIII reaching at least 0.50 IU per mL at peak, and a patient who meets this can use desmopressin to treat minor bleeds and to cover minor surgery and dental work without needing concentrate. Desmopressin is most reliable in type 1, variable in type 2A and type 2M, generally avoided in type 2B, and useless in type 3. Because the endothelial stores deplete with repeated dosing, tachyphylaxis develops, so doses are spaced at least 24 hours apart and fluid intake is restricted to prevent dilutional hyponatraemia, which is a particular risk in young children. [4] [2]
VWF concentrate is reserved for the patient who cannot use desmopressin or whose bleeding is too severe for it. It is the treatment for type 3, for type 2B, for patients who did not respond to a desmopressin test, and for major surgery and major bleeding of any type. Plasma-derived VWF and factor VIII concentrates (such as those combining VWF and factor VIII) are dosed on the VWF activity in IU per kg, and recombinant VWF (vonicog alfa) is now available and was shown in a phase 3 trial to be effective for prophylaxis and for bleeding in severe disease. The target is a VWF activity and factor VIII above 50 IU per dL for most situations, maintained with repeat dosing every 12 to 24 hours for 7 to 14 days after major surgery. [2] [12]
Recombinant VWF prophylaxis in severe von Willebrand disease (Leebeek 2022)
Phase 3 trial of recombinant von Willebrand factor for prophylaxis in patients with severe von Willebrand disease
Key finding
Recombinant VWF prophylaxis reduced the annualised bleeding rate and provided effective haemostasis for bleeding episodes, establishing recombinant VWF as a treatment option for severe disease.
Practice change
Recombinant VWF adds a plasma-free option for prophylaxis and bleeding treatment in severe von Willebrand disease, alongside plasma-derived VWF and factor VIII concentrates.
The non-replacement treatments are essential for mucosal and menstrual bleeding and are often enough on their own for mild disease. Tranexamic acid at 15 mg per kg orally three times daily (or 10 mg per kg intravenously three times daily) is effective for epistaxis, dental bleeding and heavy menstrual bleeding, and it is combined with desmopressin or concentrate for oral surgery to good effect. For the adolescent with heavy menstrual bleeding, a combined oral contraceptive pill, tranexamic acid taken during the period, or a levonorgestrel-releasing intrauterine device are all effective, and iron replacement is given for the iron deficiency anaemia that so often accompanies menorrhagia. [4] [10]
Specific Subtypes & Scenarios
Type 1 is the commonest scenario and the one a general paediatrician will meet most often. It is a partial deficiency, usually mild, and most patients respond to desmopressin and tranexamic acid and need no regular treatment at all. The priority is to confirm the diagnosis with repeat testing, to document the desmopressin response, to give the family a written plan for bleeding and procedures, and to ensure the iron status is checked in adolescent females. For dental work and minor surgery, desmopressin plus tranexamic acid usually suffices, with VWF concentrate held in reserve for a non-response. [7] [2]
Type 2B is the subtype that traps the unwary, because it presents with thrombocytopenia that can be mistaken for immune thrombocytopenia. The gain-of-function VWF binds platelets spontaneously, removing the high-molecular-weight multimers and consuming platelets, and desmopressin makes this worse by releasing more abnormal VWF and dropping the platelet count. The correct response is to recognise the increased low-dose ristocetin-induced platelet aggregation, avoid desmopressin, and treat bleeding and surgery with VWF concentrate. Pregnancy can unmask type 2B with worsening thrombocytopenia as VWF rises, which is why the platelet count is monitored through pregnancy in known type 2B. [5] [10]
Type 3 is the severe autosomal recessive form and behaves like a severe bleeding disorder. VWF is virtually absent and factor VIII is very low, so patients have severe mucocutaneous bleeding and joint and muscle bleeds, and they depend on VWF concentrate for treatment and for regular prophylaxis. The von Willebrand Disease Prophylaxis Network data showed that prophylaxis with VWF concentrate reduces bleeding in severe disease, and recombinant VWF now offers a plasma-free option. A minority of type 3 patients develop alloantibodies against infused VWF after repeated exposure, which can render concentrate ineffective and require bypassing agents, so these patients are managed in a specialist centre. [11] [12]
VWF
Complications & Pitfalls
The complications of von Willebrand disease divide into those of the bleeding and those of its treatment. The dominant bleeding complication in mild and moderate disease is iron deficiency anaemia from heavy menstrual bleeding, which is common in adolescent females and is easily overlooked. In severe type 3 disease the complications are the disabling mucocutaneous bleeding, joint bleeds and arthropathy that mirror haemophilia, and the rare but serious risk of alloantibody formation against infused VWF that makes treatment ineffective. [10] [11]
The complications of desmopressin are predictable and preventable. Because desmopressin releases stored VWF with water-retaining antidiuretic activity, it can cause dilutional hyponatraemia, which is a particular risk in young children and in patients who drink freely after the dose; fluid restriction for 24 hours after a dose is the safeguard. Tachyphylaxis develops with repeated dosing because the endothelial stores deplete, so doses are spaced at least a day apart, and in type 2B the released abnormal VWF causes transient thrombocytopenia, which is why desmopressin is generally avoided in that subtype. [4] [2]
The avoidable pitfalls are well defined. The first is missing the diagnosis because of a single normal VWF level, forgetting that blood group O lowers the level by 25 to 30 percent and that stress and oestrogen raise it; repeat testing is part of the standard when the bleeding history fits. The second is mislabelling type 2N as mild haemophilia A because the factor VIII was low and the VWF panel was not sent; the family history (autosomal, both sexes) and the factor VIII binding assay settle it. The third is giving desmopressin to a type 2B patient and watching the platelet count fall. The fourth is failing to screen the adolescent female with heavy menstrual bleeding, who is then subjected to invasive gynaecological procedures without cover. [1] [3]
Prognosis & Disposition
For the great majority with type 1 disease, the prognosis is excellent. The bleeding is mild, it is controlled by desmopressin or tranexamic acid as needed, and life expectancy and quality of life are normal. The child goes to school, plays sport, undergoes dental and surgical procedures with appropriate cover, and is rarely limited by the condition. The main determinant of outcome in type 1 is whether the diagnosis is made and a treatment plan is in place before a major bleed or procedure exposes the risk. [2] [7]
Type 3 disease carries the heaviest burden. It requires lifelong VWF concentrate, regular prophylaxis, and comprehensive care, and it carries the risks of severe bleeding, arthropathy and, in a minority, alloantibody formation. Outcomes have improved substantially with prophylaxis and with the arrival of recombinant VWF, but the disease remains a serious chronic condition that demands a specialist team. Type 2B, too, needs ongoing care because of its thrombocytopenia and its contraindication to desmopressin, and pregnancy in type 2B needs close platelet monitoring. [11] [12]
Disposition is through a comprehensive bleeding-disorder or haemophilia treatment centre that coordinates haematology, nursing, dentistry, gynaecology and social work. The family is given a written bleed management plan that states the type, the desmopressin response if known, the action to take for a bleed, and the contact for the treatment centre, and the child carries a medic alert device. School and dental plans are put in place, iron status is monitored in adolescent females, and transition to adult care is structured so that the prophylaxis regimen, the reproductive counselling and the psychosocial support are handed over without a gap. [3] [2]
Special Populations
The adolescent female is the population in whom the diagnosis is most often missed and most often matters. Heavy menstrual bleeding is the single commonest presentation of von Willebrand disease in adolescence, and it is frequently treated with iron alone or dismissed as physiological before a VWF panel is sent. Any adolescent whose menstrual loss causes iron deficiency, disrupts school or daily activities, or prompts a gynaecological referral should be screened with a VWF antigen, VWF activity and factor VIII. Management combines tranexamic acid during the period, a combined oral contraceptive or a levonorgestrel-releasing intrauterine device, iron replacement, and, for the responsive patient, desmopressin for heavy bleeding. [10] [2]
Pregnancy changes the VWF level, and understanding the change is the key to managing the pregnant patient. VWF rises through pregnancy and usually normalises by the third trimester in type 1, which protects against bleeding at delivery, but it falls back to baseline over the six weeks postpartum, when late postpartum bleeding is a real risk. Type 2B can develop worsening thrombocytopenia as the abnormal VWF rises, type 2N and type 3 do not normalise, and a multidisciplinary plan covers the mode of delivery, the threshold for neuraxial anaesthesia (which requires a haemostatic VWF level), and the postpartum surveillance. [10] [2]
[10] [5]The neonate is usually asymptomatic, but type 3 can present in the newborn with cephalohaematoma, umbilical stump bleeding or prolonged mucosal bleeding, and infants of type 2B mothers can have transient thrombocytopenia from transplacental passage of the abnormal VWF. Any neonate with unexplained mucocutaneous, umbilical or intracranial bleeding deserves a VWF panel. The transitioning adolescent needs structured handover of the diagnosis, the desmopressin response, the bleed management plan, the iron status and reproductive counselling, because the move to adult care is a high-risk period for disengagement and for missed heavy menstrual bleeding. [1] [3]
Evidence, Guidelines & Regional Differences
The cornerstone guidance is the 2008 NHLBI evidence-based diagnosis and management guidelines, the first evidence-based report on von Willebrand disease, which set out the diagnostic thresholds, the desmopressin trial, the VWF concentrate dosing and the perioperative management used worldwide. The 2013 European principles of care from the European Group on von Willebrand Disease framed the comprehensive-care model, and the Leebeek and Eikenboom 2016 New England Journal of Medicine review is the contemporary overview of the disease and its treatment. [2] [3] [1]
The prophylaxis evidence for severe disease has grown. The von Willebrand Disease Prophylaxis Network reported that regular VWF concentrate prophylaxis reduces bleeding in patients with severe type 3 and recurrent bleeding, establishing prophylaxis as a standard for the severe end of the disease. The recombinant VWF programme then delivered a plasma-free option: the phase 3 trial of recombinant VWF for prophylaxis in severe von Willebrand disease (Leebeek 2022) showed effective bleeding prevention and treatment, and recombinant VWF is now licensed alongside plasma-derived concentrates. [11] [12]
The controversies and the frontier are active. The diagnostic grey zone of a VWF level between 0.30 and 0.50 IU per mL continues to generate debate about who to label and treat, and the shift from VWF ristocetin cofactor activity to the newer glycoprotein Ib-binding assays is improving reproducibility. Extended half-life and gene therapy approaches are in development for the severe forms, and the ISTH continues to refine the classification and the bleeding assessment tools. None of these has yet displaced desmopressin, VWF concentrate and tranexamic acid as the backbone of paediatric care. [1] [7]
Exam Pearls
Von Willebrand disease is the most common inherited bleeding disorder; type 1 is about 75 to 80 percent of cases and type 3 is about 1 in a million. Von Willebrand factor has two jobs: it mediates platelet adhesion through glycoprotein Ib, which is why the disease causes mucocutaneous bleeding, and it carries and stabilises factor VIII, which is why a severe shortage drops factor VIII and allows joint bleeds. The diagnosis is built on a mucocutaneous bleeding pattern with a normal platelet count and a normal or near-normal coagulation screen, confirmed by a low VWF antigen, activity and factor VIII. [1] [5]
The classification is the high-yield structure. Type 1 is a partial quantitative deficiency with normal multimers and is usually desmopressin-responsive. Type 2 is a qualitative defect: 2A loses the high-molecular-weight multimers, 2B is a gain-of-function with thrombocytopenia and a contraindication to desmopressin, 2M has normal multimers but reduced function, and 2N has reduced factor VIII binding and mimics haemophilia A. Type 3 is virtual absence, severe and autosomal recessive, and does not respond to desmopressin. An activity-to-antigen ratio below about 0.6 points to a type 2 qualitative defect. [5] [2]
The treatment ladder is the other high-yield structure. Desmopressin 0.3 micrograms per kg intravenously over 30 minutes releases stored VWF and factor VIII with a 2 to 4 fold rise, and an adequate response is activity reaching at least 0.50 IU per mL; it is first-line for type 1, generally avoided in type 2B, and useless in type 3. VWF concentrate (plasma-derived or recombinant) is used for type 3, type 2B, non-responders, and major bleeding or surgery, targeting activity above 50 IU per dL. Tranexamic acid 15 mg per kg orally three times daily covers mucosal and menstrual bleeding. [4] [2]
The one-liners an examiner rewards: blood group O lowers VWF by 25 to 30 percent, so a single normal level never excludes the disease; type 2N is the autosomal mimic of mild haemophilia A, separated by the factor VIII binding assay; heavy menstrual bleeding is the commonest presentation in adolescent females and every such patient should be screened; desmopressin causes hyponatraemia through water retention, so restrict fluids for 24 hours; and recombinant VWF is now licensed for severe disease following the 2022 phase 3 prophylaxis trial. [1] [12]
References
- [1]Leebeek FW, Eikenboom JC Von Willebrand's Disease. N Engl J Med, 2016.PMID 27959741
- [2]Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report. Haemophilia, 2008.PMID 18315614
- [3]Castaman G, Goodeve A, Eikenboom J, European Group on von Willebrand Disease Principles of care for the diagnosis and treatment of von Willebrand disease. Haematologica, 2013.PMID 23633542
- [4]Mannucci PM Treatment of von Willebrand's Disease. N Engl J Med, 2004.PMID 15306670
- [5]Sadler JE A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost, 1994.PMID 8052974
- [6]Rodeghiero F, Castaman G, Dini E Epidemiological investigation of the prevalence of von Willebrand's disease. Blood, 1987.PMID 3492222
- [7]Sadler JE Von Willebrand disease type 1: a diagnosis in search of a disease. Blood, 2003.PMID 12411289
- [8]Rodeghiero F, Tosetto A, Abshire T, Arnold DM, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost, 2010.PMID 20626619
- [9]Tosetto A, Castaman G, Rodeghiero F Assessing bleeding in von Willebrand disease with bleeding score. Blood Rev, 2007.PMID 16774804
- [10]Mannucci PM, Federici AB, James AH, Kessler CM von Willebrand disease in the 21st century: current approaches and new challenges. Haemophilia, 2009.PMID 19624761
- [11]Abshire TC, Federici AB, Alvarez MT, Bowen J, et al. Prophylaxis in severe forms of von Willebrand's disease: results from the von Willebrand Disease Prophylaxis Network (VWD PN). Haemophilia, 2013.PMID 22823000
- [12]Leebeek FWG, Peyvandi F, Escobar M, Tiede A, et al. Recombinant von Willebrand factor prophylaxis in patients with severe von Willebrand disease: phase 3 trial results. Blood, 2022.PMID 35439298