Adult-Onset Still's Disease
Adult-onset Still's disease (AOSD) is a rare systemic autoinflammatory disorder characterized by the classic triad of qu... MRCP exam preparation.
What matters first
Adult-onset Still's disease (AOSD) is a rare systemic autoinflammatory disorder characterized by the classic triad of qu... MRCP exam preparation.
Macrophage activation syndrome (MAS) - ferritin drop, cytopenias, coagulopathy
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Macrophage activation syndrome (MAS) - ferritin drop, cytopenias, coagulopathy
- Severe hepatic involvement with liver failure
- Cardiac tamponade from pericarditis
- Respiratory failure from ARDS or pleuritis
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- Systemic Lupus Erythematosus
- Sepsis and Infection Syndromes
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Adult-Onset Still's Disease
1. Overview
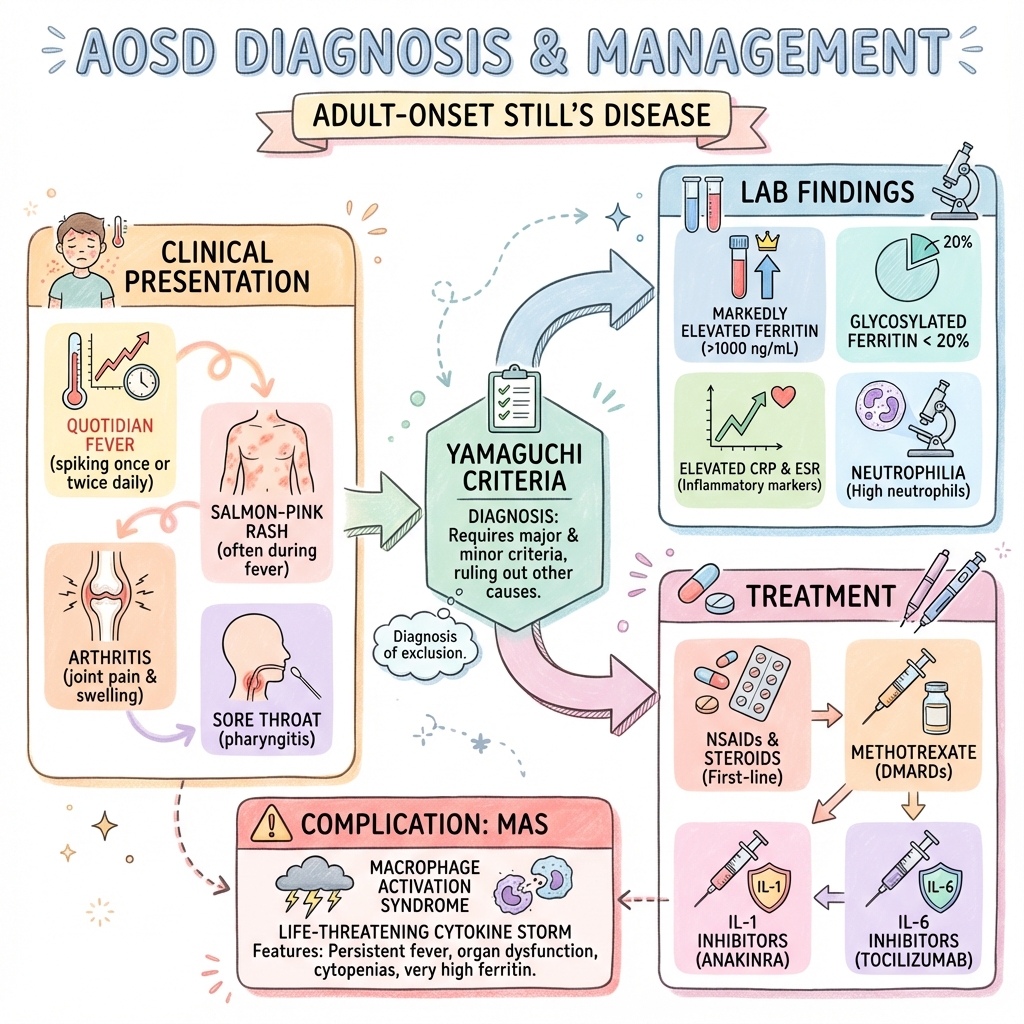
Adult-onset Still's disease (AOSD) is a rare systemic autoinflammatory disorder characterized by the classic triad of quotidian (once-daily) spiking fevers, evanescent salmon-pink rash, and arthritis or arthralgia. First described by George Still in children in 1897, the adult variant was recognized as a distinct entity in the 1970s. AOSD represents the adult counterpart of systemic juvenile idiopathic arthritis (sJIA), sharing similar clinical and immunological features. [1]
The condition is distinguished from autoimmune diseases by the absence of autoantibodies and the central role of innate immune system dysregulation rather than adaptive immunity. The pathogenesis involves overproduction of proinflammatory cytokines, particularly interleukin-1β (IL-1β), interleukin-6 (IL-6), and interleukin-18 (IL-18), leading to systemic inflammation. [2] This understanding has revolutionized treatment, with biologic therapies targeting these cytokines demonstrating superior efficacy compared to traditional immunosuppression.
AOSD remains a diagnosis of exclusion, requiring careful elimination of infectious, malignant, and other autoimmune causes. The Yamaguchi criteria, developed in 1992, remain the most widely used diagnostic framework, requiring at least 5 criteria including at least 2 major criteria after exclusion of infections, malignancies, and other rheumatic diseases. [3] Laboratory findings of markedly elevated ferritin (often > 1000 ng/mL) with low glycosylated ferritin fraction (less than 20%) have emerged as highly specific markers. [4] Macrophage activation syndrome (MAS), occurring in 10-15% of patients, represents a life-threatening complication requiring urgent recognition and treatment. [5]
2. Epidemiology
AOSD is a rare disease with an estimated annual incidence of 0.16 to 0.4 per 100,000 population in European studies, with slightly higher rates reported in Japan (0.34 per 100,000). [6,7] The true prevalence may be underestimated due to underdiagnosis of this challenging condition.
Demographics
The disease demonstrates a characteristic bimodal age distribution with peaks at 15-25 years and 36-46 years, though onset can occur at any age in adulthood. [8] Some studies report a third peak in the elderly (> 60 years). There is a slight female predominance with a female-to-male ratio of approximately 1.5:1 in most series. [1,6]
No clear ethnic predisposition has been established, though the disease has been reported worldwide across all ethnic groups. Asian populations, particularly Japanese and Chinese, show slightly higher incidence rates compared to Western populations, though this may reflect reporting bias or genetic factors. [9]
Risk Factors
Unlike many autoimmune diseases, specific environmental or genetic risk factors for AOSD remain poorly defined. Some studies have suggested associations with:
- Genetic factors: HLA-B1801, HLA-DR4, and HLA-DRB115 alleles have been reported in some populations, though associations are inconsistent across ethnic groups. [10]
- Infectious triggers: Viral infections (EBV, CMV, parvovirus B19, rubella) and bacterial infections have been proposed as potential triggers, though causality remains unproven. [11]
- Seasonality: Some case series suggest increased onset during spring and summer months, potentially supporting an infectious or environmental trigger. [12]
Epidemiological Statistics
| Statistic | Value | Reference |
|---|---|---|
| Annual incidence (Europe) | 0.16-0.4 per 100,000 | [6] |
| Annual incidence (Japan) | 0.34 per 100,000 | [7] |
| Female:male ratio | 1.5:1 | [1] |
| Age peak (first) | 15-25 years | [8] |
| Age peak (second) | 36-46 years | [8] |
| MAS complication rate | 10-15% | [5] |
| Monocyclic pattern | 30-40% | [13] |
| Polycyclic pattern | 30-40% | [13] |
| Chronic articular pattern | 20-30% | [13] |
3. Aetiology and Pathophysiology
Etiology
The precise etiology of AOSD remains unknown. Current evidence suggests that in genetically predisposed individuals, environmental triggers (likely infectious agents) initiate an inappropriate and sustained innate immune response. However, no single infectious agent has been consistently identified, and direct causality has not been established. [11]
Pathophysiology: Autoinflammatory, Not Autoimmune
AOSD is fundamentally an autoinflammatory disorder characterized by dysregulation of the innate immune system, distinguishing it from classical autoimmune diseases that involve adaptive immunity and autoantibody production. [2]
Innate Immune Dysregulation
The core pathophysiological mechanism involves hyperactivation of innate immune cells (neutrophils, macrophages, monocytes, and natural killer cells) with excessive production of proinflammatory cytokines. This creates a self-perpetuating inflammatory cascade without the adaptive immune involvement typical of autoimmune diseases. [14]
Molecular Pathophysiology: The Cytokine Storm
Three key cytokines drive the pathogenesis of AOSD, each contributing distinct clinical features:
1. Interleukin-1β (IL-1β)
- Produced by activated macrophages and neutrophils via NLRP3 inflammasome activation
- Drives fever and systemic inflammatory response
- Induces production of acute phase reactants
- Stimulates neutrophil recruitment and activation
- Central to pathogenesis: explains dramatic response to IL-1 blockade with anakinra
- Elevated in serum and synovial fluid of AOSD patients [15]
2. Interleukin-6 (IL-6)
- Produced by activated macrophages, fibroblasts, and endothelial cells
- Drives acute phase response, particularly ferritin production
- Responsible for anemia of chronic disease
- Induces thrombocytosis
- Contributes to joint destruction in chronic articular disease
- Levels correlate with disease activity [16]
3. Interleukin-18 (IL-18)
- Belongs to IL-1 superfamily, produced by activated macrophages
- Extremely elevated in AOSD (often 10-20 fold above normal)
- Induces interferon-γ production from NK cells and T cells
- Levels correlate with disease activity and risk of MAS
- Potential biomarker for monitoring and predicting complications [17]
The Inflammasome Connection
The NLRP3 (NOD-like receptor protein 3) inflammasome plays a central role:
- Pattern recognition receptor complex activated by damage-associated molecular patterns (DAMPs)
- Triggers caspase-1 activation
- Processes pro-IL-1β to active IL-1β
- Processes pro-IL-18 to active IL-18
- Represents therapeutic target for inflammasome inhibitors [18]
Neutrophil Activation
Neutrophils are dramatically activated in AOSD:
- Marked neutrophilia (often > 80% of white blood cells)
- Production of S100A8/A9 (calprotectin) and S100A12 proteins
- Release of neutrophil extracellular traps (NETs)
- S100 proteins serve as DAMPs, further activating inflammasomes
- Calprotectin levels correlate with disease activity [19]
Ferritin Biology
The characteristic ferritin elevation in AOSD has specific mechanisms:
- IL-6 and IL-1β induce ferritin production by hepatocytes and macrophages
- Ferritin acts as both acute phase reactant and cytokine (immunomodulatory properties)
- Glycosylated ferritin fraction decreases in acute inflammation
- Normal glycosylated fraction: 50-80%
- AOSD glycosylated fraction: typically less than 20%
- Mechanism: rapid production of ferritin in acute inflammation overwhelms glycosylation machinery
- Glycosylated fraction less than 20% has 70% sensitivity and 83% specificity for AOSD [4]
Why Absence of Autoantibodies?
Unlike autoimmune diseases such as SLE or rheumatoid arthritis, AOSD is characterized by:
- Negative ANA (antinuclear antibody)
- Negative RF (rheumatoid factor)
- Absence of disease-specific autoantibodies
- Minimal adaptive immune involvement
- Innate immune cell predominance
This reflects the fundamental difference between autoinflammatory (innate immune) and autoimmune (adaptive immune) mechanisms. [2]
4. Clinical Presentation
AOSD presents with a characteristic constellation of clinical features, though individual patients may show significant variability. The classic triad of quotidian fever, evanescent rash, and arthritis is present in approximately 60% of patients at presentation. [20]
Cardinal Features
Fever (> 95% of patients)
The fever pattern in AOSD is highly characteristic and often provides an important diagnostic clue:
- Quotidian fever: Single daily spike, typically in the late afternoon or evening (4-8 PM)
- Temperature: Usually > 39°C (102.2°F), often reaching 40°C or higher
- Pattern: Rapid rise to peak, followed by return to normal or subnormal temperature
- Duration: Each fever spike lasts several hours
- Associated symptoms: Patients often feel systemically unwell during fever spikes, with improvement between episodes
- Response to antipyretics: Variable, often incomplete response
Clinical Pearl: Patients may appear relatively well between fever spikes, leading to initial dismissal of symptoms. Documented temperature charts showing the quotidian pattern are diagnostically valuable. [1]
Rash (75-95% of patients)
The AOSD rash has distinctive characteristics:
- Appearance: Salmon-pink or faint pink macules and maculopapular lesions
- Distribution: Trunk and proximal extremities most common; face, palms, soles can be involved
- Timing: Typically appears during fever spikes, fades when temperature normalizes (evanescent)
- Duration: Individual lesions last hours
- Koebner phenomenon: Rash may be induced by scratching, rubbing, or trauma (positive Koebner sign)
- Pruritus: Typically non-pruritic or minimally pruritic
- Dermographism: Often present
Diagnostic Challenge: The evanescent nature means the rash may not be visible during clinic visits. Patients should be examined during febrile episodes or asked to photograph the rash when it appears. Skin biopsy shows non-specific perivascular neutrophilic infiltration. [20]
Arthritis/Arthralgia (75-100% of patients)
Joint involvement is nearly universal but varies in pattern:
- Timing: May precede, accompany, or follow systemic features
- Pattern: Typically polyarticular, symmetric
- Joints involved:
- Knees (most common)
- Wrists and ankles
- Shoulders and hips
- Small joints of hands and feet
- Cervical spine
- Temporomandibular joints
- Ankylosis of carpal and tarsal bones is characteristic in chronic disease
- Character: Initially often arthralgia without synovitis; progresses to true arthritis
- Severity: Ranges from mild stiffness to severe destructive arthropathy
Chronic Articular Pattern: 20-30% of patients develop chronic destructive arthritis as the dominant feature, with less prominent systemic symptoms. This pattern has worse functional outcomes. [13]
Other Clinical Features
| Feature | Frequency | Characteristics | Clinical Significance |
|---|---|---|---|
| Sore throat | 50-90% | Pharyngitis without exudate; early symptom | May mimic infectious pharyngitis |
| Lymphadenopathy | 40-75% | Generalized; cervical most common | Requires exclusion of lymphoma |
| Hepatomegaly | 40-55% | Mild to moderate enlargement | Check transaminases |
| Splenomegaly | 25-65% | Usually mild | Associated with higher disease activity |
| Serositis | 20-40% | Pleuritis, pericarditis | Can cause tamponade (rare) |
| Myalgia | 60-85% | Generalized muscle pain | Often accompanies fever |
| Weight loss | 40-60% | Significant (> 5 kg) | Reflects systemic inflammation |
| Abdominal pain | 15-50% | Non-specific; may relate to hepatosplenomegaly or serositis | Exclude other causes |
Rare but Recognized Manifestations
- Cardiac: Myocarditis, pericardial tamponade, valvular disease
- Pulmonary: Acute respiratory distress syndrome (ARDS), pulmonary hypertension, interstitial lung disease
- Neurological: Aseptic meningitis, seizures (rare)
- Renal: Amyloidosis (chronic complication), glomerulonephritis (rare)
- Hematological: Thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC)
5. Differential Diagnosis
AOSD is a diagnosis of exclusion. The differential diagnosis is broad and includes infections, malignancies, and other rheumatological conditions. Systematic exclusion is essential. [3]
Priority Differentials
1. Infectious Diseases (Must Not Miss)
Sepsis and Bacteremia
- Key distinguishing features: Continuous fever (not quotidian), hypotension, positive blood cultures
- Investigation: Blood cultures (ideally before antibiotics), lactate, procalcitonin
- Clinical context: Presence of source (pneumonia, UTI, cellulitis)
Endocarditis
- Key distinguishing features: New cardiac murmur, embolic phenomena, positive blood cultures
- Investigation: Echocardiography (TEE more sensitive), serial blood cultures
- Clinical context: Risk factors (IV drug use, prosthetic valves)
Viral Infections
- EBV, CMV, HIV, parvovirus B19, hepatitis viruses
- Key distinguishing features: Specific serological profiles, lymphocytosis (not neutrophilia)
- Investigation: Viral serology, PCR where appropriate
Tuberculosis
- Key distinguishing features: Night sweats, pulmonary infiltrates, positive IGRA/tuberculin test
- Investigation: Chest X-ray, IGRA, culture
- Clinical context: Endemic areas, immunosuppression, constitutional symptoms
2. Malignancy
Lymphoma (Hodgkin and Non-Hodgkin)
- Key distinguishing features: Persistent lymphadenopathy, B-symptoms, mass lesions
- Investigation: CT imaging, lymph node biopsy, bone marrow biopsy
- Clinical pearl: Pel-Ebstein fever pattern in Hodgkin lymphoma vs quotidian in AOSD
Leukemia
- Key distinguishing features: Abnormal blood film, blasts on differential
- Investigation: Blood film, flow cytometry, bone marrow biopsy
Solid Tumors with Paraneoplastic Fever
- Renal cell carcinoma, hepatocellular carcinoma particularly
- Investigation: CT abdomen/pelvis, tumor markers
3. Other Rheumatological Conditions
Systemic Lupus Erythematosus (SLE)
- Key distinguishing features: Positive ANA, anti-dsDNA; malar rash; renal involvement
- Investigation: ANA, ENA panel, complement levels, urinalysis
- Clinical context: More common in young women; photosensitive rash
Polyarteritis Nodosa and Other Vasculitides
- Key distinguishing features: ANCA positivity, specific organ involvement patterns
- Investigation: ANCA, biopsy, angiography
- Clinical context: Renal, neurological involvement
Reactive Arthritis
- Key distinguishing features: Asymmetric oligoarthritis, enthesitis, conjunctivitis, urethritis
- Investigation: HLA-B27, stool/urine cultures
- Clinical context: Recent GI or GU infection
Familial Mediterranean Fever (FMF) and Other Hereditary Periodic Fevers
- Key distinguishing features: Episodic fever (24-72 hours), family history, ethnicity
- Investigation: Genetic testing (MEFV mutations), response to colchicine
- Clinical context: Mediterranean ancestry, childhood onset more common
4. Drug Reactions
Drug-Induced Hypersensitivity (DRESS/DIHS)
- Key distinguishing features: Recent drug exposure, eosinophilia, facial edema
- Investigation: Eosinophil count, discontinuation of suspect drug
- Clinical context: Temporal relationship with medication
Differential Diagnosis Comparison Table
| Condition | Fever Pattern | Rash | Ferritin | Autoantibodies | Key Distinguisher |
|---|---|---|---|---|---|
| AOSD | Quotidian | Salmon-pink, evanescent | Very high (> 1000) | Negative | Low glycosylated ferritin |
| Sepsis | Continuous | Variable | Elevated | N/A | Positive cultures, hypotension |
| Lymphoma | Variable/Pel-Ebstein | Variable | Elevated | Negative | Lymphadenopathy, biopsy |
| SLE | Variable | Malar, photosensitive | Moderate | Positive ANA, dsDNA | Renal involvement |
| FMF | Episodic (24-72h) | Erysipelas-like | Moderate | Negative | Family history, MEFV mutation |
| Viral infection | Variable | Variable | Moderate | Variable | Lymphocytosis, viral serology |
6. Investigations
The Yamaguchi Criteria (1992)
The Yamaguchi criteria remain the most widely used diagnostic classification for AOSD. Diagnosis requires at least 5 criteria including at least 2 major criteria, after exclusion of infections, malignancies, and other rheumatic diseases. [3]
Major Criteria
- Fever ≥39°C lasting ≥1 week
- Arthralgia or arthritis lasting ≥2 weeks
- Typical rash (salmon-pink, evanescent, macular)
- Leukocytosis ≥10,000/μL with ≥80% granulocytes
Minor Criteria
- Sore throat
- Lymphadenopathy and/or splenomegaly
- Liver enzyme abnormalities (AST or ALT elevation)
- Negative rheumatoid factor (RF) and negative antinuclear antibody (ANA)
Sensitivity and Specificity: The Yamaguchi criteria have sensitivity of 96% and specificity of 92% for AOSD when applied correctly with appropriate exclusions. [3]
Other Diagnostic Criteria
Fautrel Criteria (2002): Alternative classification system
- Major criteria: Spiking fever ≥39°C, arthralgia, transient erythema, pharyngitis, neutrophilia ≥80%, glycosylated ferritin ≤20%
- Minor criteria: Maculopapular rash, leukocytosis ≥10,000/μL
- Requires 4 major or 3 major + 2 minor
- Incorporates glycosylated ferritin as major criterion
- Sensitivity 81%, specificity 98% [4]
Laboratory Investigations
First-Line Investigations
Complete Blood Count
- Leukocytosis: Typically 15,000-40,000/μL (can exceed 50,000/μL)
- Neutrophil predominance: Usually > 80% granulocytes
- Anemia: Normocytic normochromic anemia of chronic disease (common)
- Thrombocytosis: Mild to moderate elevation (reactive)
- Note: Leukopenia or thrombocytopenia should raise concern for MAS
Inflammatory Markers
- ESR: Markedly elevated (often > 100 mm/hr)
- CRP: Markedly elevated (often > 100 mg/L)
- Both correlate with disease activity
Ferritin and Glycosylated Ferritin
- Ferritin: Typically > 1000 ng/mL; often > 3000-5000 ng/mL; can exceed 50,000 ng/mL
- Five-fold elevation above upper limit of normal has 80% sensitivity for AOSD
- Glycosylated ferritin fraction: less than 20% is highly specific (83%) for AOSD [4]
- Normal glycosylated fraction: 50-80%
- Mechanism: Rapid ferritin production in acute inflammation saturates glycosylation capacity
- Clinical utility: Helps distinguish AOSD from other causes of hyperferritinemia (infection, malignancy, hemophagocytic syndromes)
Liver Function Tests
- Transaminases: Elevated in 50-75% (usually less than 5x ULN)
- Alkaline phosphatase: May be elevated
- Bilirubin: Usually normal; elevation suggests severe disease or MAS
- Pattern: Hepatocellular > cholestatic
Autoantibodies (for exclusion)
- ANA: Negative (positive ANA should prompt reconsideration of SLE)
- RF: Negative
- ANCA: Negative
- Anti-CCP: Negative
Second-Line and Specialized Investigations
Cytokine Levels (Research/Specialized Centers)
- IL-18: Markedly elevated (10-20 fold); correlates with disease activity and MAS risk [17]
- IL-6: Elevated; correlates with disease activity
- IL-1β: Elevated during active disease
- Not routinely available but helpful when accessible
Imaging
Plain Radiographs
- Early disease: Often normal or non-specific soft tissue swelling
- Chronic disease:
- Carpal and tarsal ankylosis (highly characteristic)
- Joint space narrowing
- Erosions (less prominent than rheumatoid arthritis)
Ultrasound/MRI
- Utility: Detect early synovitis, effusions
- MRI: Useful for assessing bone marrow edema, early erosions
CT Chest/Abdomen/Pelvis
- Purpose: Exclude infection, malignancy
- Findings in AOSD: Lymphadenopathy, hepatosplenomegaly, serositis
PET-CT
- Utility: High metabolic activity in lymph nodes, spleen, bone marrow
- Can mimic lymphoma: Requires careful interpretation
- Useful: When malignancy exclusion is challenging
Exclusionary Investigations
Essential to exclude alternative diagnoses:
| Investigation | Purpose |
|---|---|
| Blood cultures (×3 sets) | Exclude bacteremia, endocarditis |
| Viral serology | Exclude EBV, CMV, HIV, hepatitis |
| Tuberculin test/IGRA | Exclude tuberculosis |
| Procalcitonin | Bacterial infection (low in AOSD) |
| Lymph node biopsy | Exclude lymphoma if persistent lymphadenopathy |
| Bone marrow biopsy | If atypical features, cytopenias, or MAS concern |
| Echocardiography | Assess for endocarditis, pericardial effusion |
Monitoring Investigations During Treatment
- Ferritin: Follows disease activity; use for monitoring response
- CRP/ESR: Track inflammation
- CBC: Monitor for cytopenias suggesting MAS
- LFTs: Monitor for drug hepatotoxicity and disease activity
- IL-18 (if available): Predicts flares and MAS risk
7. Management
Treatment of AOSD has evolved significantly with the introduction of biologic therapies targeting key cytokines. The treatment approach is tailored to disease severity and pattern (systemic vs articular predominance). [1]
Treatment Algorithm
Mild Disease (Predominantly Articular, No Organ Involvement)
- NSAIDs ± low-dose corticosteroids
- If inadequate response: Add methotrexate or other DMARD
- If refractory: Consider biologic therapy
Moderate to Severe Disease (Significant Systemic Features)
- Corticosteroids (prednisolone 0.5-1 mg/kg/day)
- Early addition of steroid-sparing agent (methotrexate) or biologic
- If refractory: Switch to alternative biologic
Life-Threatening Disease (MAS, Organ Failure)
- High-dose IV methylprednisolone (1000 mg/day × 3-5 days)
- Anakinra or other IL-1 inhibitor
- Consider cyclosporine for MAS
- ICU support as needed
First-Line Therapies
Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
Indications: Mild disease, predominantly articular symptoms Mechanism: COX inhibition reduces prostaglandin synthesis Efficacy: Effective in less than 20% as monotherapy; most require additional treatment Agents:
- Indomethacin 100-200 mg/day in divided doses (historically most used)
- Naproxen 500-1000 mg/day in divided doses
- Ibuprofen 1200-2400 mg/day in divided doses
Limitations:
- Hepatotoxicity risk increased in AOSD (baseline liver involvement)
- Monitoring: LFTs, renal function
- Avoid in severe disease, hepatic involvement
Corticosteroids
Indications: Moderate to severe disease; first-line for systemic features Mechanism: Broad anti-inflammatory and immunosuppressive effects Efficacy: 60-80% achieve initial response Dosing:
- Prednisolone 0.5-1 mg/kg/day orally (moderate disease)
- Methylprednisolone 500-1000 mg IV daily × 3-5 days (severe disease/MAS)
- Taper once disease controlled (typically over months)
Response: Usually within days to 1-2 weeks Limitations:
- 25% are steroid-resistant
- 60-70% cannot taper below 10 mg/day without relapse (steroid-dependent)
- Significant side effects with long-term use
- Early introduction of steroid-sparing agents recommended
Disease-Modifying Anti-Rheumatic Drugs (DMARDs)
Methotrexate
Indications: Steroid-sparing agent; particularly for articular disease Mechanism: Antifolate; reduces inflammation Efficacy: 60-70% show benefit, particularly in chronic articular pattern Dosing: 15-25 mg weekly (oral or subcutaneous) Adjunct: Folic acid 5 mg weekly (on different day) Monitoring: CBC, LFTs, renal function at baseline and regularly (typically monthly initially) Onset: 6-12 weeks Limitations: Less effective for systemic features; requires folate supplementation; hepatotoxic; contraindicated in pregnancy
Other DMARDs (Less Commonly Used)
- Azathioprine: 1-2.5 mg/kg/day; steroid-sparing
- Leflunomide: 10-20 mg/day; alternative to methotrexate
- Cyclosporine: Particularly for MAS; 2-5 mg/kg/day
- Hydroxychloroquine: Limited efficacy in AOSD
Biologic Therapies
Biologic therapies targeting IL-1 and IL-6 have transformed AOSD management, demonstrating superior efficacy compared to traditional immunosuppression. [21]
IL-1 Inhibitors
Anakinra (IL-1 Receptor Antagonist)
- Mechanism: Recombinant IL-1 receptor antagonist; blocks IL-1α and IL-1β signaling
- Efficacy:
- Dramatic response in systemic symptoms (fever, rash) within 24-48 hours
- "Complete response rates: 60-70%"
- Particularly effective for systemic features
- Beneficial in MAS
- Dosing: 100 mg subcutaneous daily
- Onset: Rapid (hours to days for fever/rash; weeks for articular symptoms)
- Evidence: Multiple case series and cohort studies; no large RCTs in AOSD specifically [21]
- Advantages:
- Rapid onset
- Short half-life (allows quick discontinuation if needed)
- Effective steroid-sparing agent
- Safe in pregnancy (category B)
- Limitations:
- Daily injection
- Injection site reactions (common, usually mild)
- Articular response may be less complete than systemic response
- Monitoring: Baseline and periodic CBC, infection surveillance
Canakinumab (IL-1β Monoclonal Antibody)
- Mechanism: Fully human monoclonal antibody targeting IL-1β
- Efficacy: Similar to anakinra; may have better articular response
- Dosing: 4 mg/kg (maximum 300 mg) subcutaneous every 4-8 weeks
- Onset: Days to weeks
- Evidence: RCT data in systemic JIA; case series in AOSD [22]
- Advantages:
- Less frequent dosing (monthly)
- May have superior articular efficacy
- Limitations:
- More expensive
- Longer half-life (less flexibility)
- Monitoring: As for anakinra
Rilonacept: IL-1 trap (binds IL-1α and IL-1β); less commonly used; weekly subcutaneous injection
IL-6 Inhibitors
Tocilizumab (IL-6 Receptor Antibody)
- Mechanism: Humanized monoclonal antibody blocking IL-6 receptor
- Efficacy:
- Effective for both systemic and articular features
- "Complete response rates: 60-80%"
- Particularly effective for chronic articular disease
- Normalizes ferritin levels
- Dosing:
- 8 mg/kg IV every 4 weeks, or
- 162 mg subcutaneous weekly
- Onset: 1-4 weeks
- Evidence: Multiple cohort studies and case series; high-quality evidence in systemic JIA [23]
- Advantages:
- Effective for both systemic and articular manifestations
- Less frequent dosing (monthly IV)
- Excellent for chronic articular pattern
- Limitations:
- Risk of infection
- Lipid abnormalities (monitor lipid profile)
- Neutropenia risk
- Masks inflammatory markers (CRP suppression may complicate infection diagnosis)
- Monitoring:
- "Baseline: TB screening, hepatitis B/C, CBC, LFTs, lipid profile"
- "Periodic: CBC (neutropenia risk), LFTs, lipid profile"
Sarilumab: Alternative IL-6 receptor antibody; similar efficacy; 200 mg subcutaneous every 2 weeks
Other Biologics
TNF Inhibitors (Etanercept, Infliximab, Adalimumab)
- Efficacy: Variable and generally disappointing in AOSD (unlike rheumatoid arthritis)
- Response rates: 30-50% (lower than IL-1/IL-6 inhibitors)
- Current role: Not first-line; may consider if IL-1/IL-6 inhibitors unavailable or failed
- Evidence: Case series; no RCT data [24]
JAK Inhibitors (Tofacitinib, Baricitinib)
- Emerging data: Case reports suggest potential efficacy
- Mechanism: Inhibit Janus kinases involved in cytokine signaling
- Role: Investigational; limited data
Choosing Between IL-1 and IL-6 Inhibitors
| Feature | IL-1 Inhibitor (Anakinra) | IL-6 Inhibitor (Tocilizumab) |
|---|---|---|
| Systemic symptoms | Very rapid response (24-48h) | Effective (1-2 weeks) |
| Articular symptoms | Good but may be incomplete | Excellent, especially chronic arthritis |
| MAS | Effective, rapid | Less data, may mask inflammation |
| Dosing | Daily injection | Monthly IV or weekly SC |
| Onset | Hours to days | Days to weeks |
| Cost | Lower (anakinra) | Higher |
| Preferred for | Acute systemic disease, MAS | Chronic articular disease |
Expert Recommendations:
- Predominantly systemic, acute: Anakinra first-line
- Chronic articular pattern: Tocilizumab may be preferred
- MAS: Anakinra (rapid onset, safe)
- Switch to alternative if inadequate response at 3 months
Management of Macrophage Activation Syndrome (MAS)
MAS is a life-threatening complication occurring in 10-15% of AOSD patients, characterized by hemophagocytosis and cytokine storm. [5]
Clinical Features of MAS:
- Continuous fever (loss of quotidian pattern)
- Altered mental status
- Hepatosplenomegaly
- Purpura, bleeding
- Hypotension, shock
Laboratory Features (HLH-2004 Criteria):
- Falling ferritin (paradoxically, as acute phase response fails)
- Cytopenias (at least 2 lineages): anemia, thrombocytopenia, neutropenia
- Hypertriglyceridemia (> 265 mg/dL)
- Hypofibrinogenemia (less than 150 mg/dL)
- Elevated transaminases
- Elevated LDH
- Elevated soluble IL-2 receptor
- Hemophagocytosis on bone marrow biopsy
Management:
- High-dose corticosteroids: Methylprednisolone 1000 mg IV daily × 3-5 days
- Anakinra: 100 mg SC daily or higher doses (up to 2-4 mg/kg/day)
- Cyclosporine: 2-5 mg/kg/day (particularly effective for MAS)
- Etoposide: Consider in refractory cases (chemotherapy protocol as per HLH)
- Supportive care: ICU monitoring, transfusion support, treat coagulopathy
Prognosis: Mortality 10-20% despite treatment; early recognition and aggressive therapy essential
Treatment of Refractory Disease
Definition: Inadequate response to corticosteroids plus at least one DMARD or biologic
Strategies:
- Switch biologics: IL-1 inhibitor ↔ IL-6 inhibitor
- Combination therapy: Biologic + methotrexate
- Higher doses: Anakinra up to 300 mg daily (off-label)
- Alternative agents:
- Cyclosporine (particularly if MAS features)
- Intravenous immunoglobulin (IVIG)
- JAK inhibitors (investigational)
Monitoring During Treatment
| Parameter | Frequency | Purpose |
|---|---|---|
| Clinical assessment | Every visit | Disease activity, treatment response |
| ESR, CRP | Monthly initially, then 3-monthly | Inflammatory activity |
| Ferritin | Monthly initially, then as needed | Disease activity marker |
| CBC | Monthly (especially on biologics/DMARDs) | Cytopenias, MAS screening |
| LFTs | Monthly initially, then 3-monthly | Hepatotoxicity, disease activity |
| Renal function | 3-6 monthly | Baseline, amyloidosis screening |
| Lipid profile | 3-6 monthly (on tocilizumab) | IL-6 inhibitor effect |
| Infection surveillance | Ongoing | Immunosuppression risk |
8. Complications
Macrophage Activation Syndrome (MAS)
Incidence: 10-15% of AOSD patients [5] Timing: Can occur at presentation or during disease course Trigger: Disease flare, infection, medication changes Pathophysiology: Uncontrolled activation of macrophages and T cells; hemophagocytosis; cytokine storm Clinical features: Described in Management section above Management: High-dose steroids, anakinra, cyclosporine Prognosis: Mortality 10-20%
Red Flags for MAS:
- Continuous fever replacing quotidian pattern
- Falling platelet count
- Paradoxically falling ferritin (but still elevated)
- Rising liver enzymes
- Altered mental status
- Coagulopathy
Chronic Destructive Arthritis
Incidence: 20-30% develop chronic articular pattern [13] Joints affected: Wrists, ankles, knees; carpal and tarsal ankylosis characteristic Consequences: Joint destruction, functional impairment Radiographic findings:
- Carpal ankylosis (highly characteristic)
- Tarsal ankylosis
- Joint space narrowing
- Erosions (less than RA) Prevention: Early aggressive treatment, particularly with IL-6 inhibitors Management: DMARDs, biologics (tocilizumab particularly effective), physiotherapy, surgical intervention for severe cases
Secondary Amyloidosis (AA Amyloidosis)
Incidence: Rare in modern era with effective treatment; historically up to 5-10% Pathophysiology: Chronic inflammation leads to deposition of serum amyloid A protein Organs affected: Kidneys (most common), GI tract, liver, heart Clinical features: Proteinuria, nephrotic syndrome, renal failure Diagnosis: Renal biopsy showing amyloid deposits (Congo red stain) Prevention: Tight control of inflammation Prognosis: Poor if renal involvement advanced; dialysis-dependent
Cardiac Complications
Pericarditis: 20-40% prevalence
- Usually mild
- Rarely progresses to tamponade
- Management: NSAIDs, corticosteroids, drainage if tamponade
Myocarditis: Rare but serious
- Presents with heart failure, arrhythmias
- Requires aggressive immunosuppression
Valvular disease: Rare; usually mild
Pulmonary Complications
Pleuritis/Pleural effusion: 20-30% Acute respiratory distress syndrome (ARDS): Rare, life-threatening Interstitial lung disease: Uncommon Pulmonary hypertension: Rare
Hepatic Complications
Transaminitis: Common (50-75%), usually mild Acute liver failure: Rare but described Monitoring: Regular LFTs essential
Hematological Complications
Disseminated Intravascular Coagulation (DIC): Rare, associated with MAS Thrombotic thrombocytopenic purpura (TTP): Very rare case reports
Infectious Complications
Risk: Increased due to immunosuppression (corticosteroids, biologics) Infections: Bacterial, opportunistic (TB reactivation, fungal, PCP) Prevention:
- TB screening before biologics
- Pneumococcal, influenza vaccination
- PCP prophylaxis if high-dose steroids
- Infection surveillance
9. Prognosis and Disease Patterns
AOSD demonstrates three distinct disease course patterns with different prognoses. [13]
Disease Patterns
| Pattern | Frequency | Description | Prognosis |
|---|---|---|---|
| Monocyclic | 30-40% | Single episode followed by complete remission | Excellent; full recovery |
| Polycyclic | 30-40% | Recurrent flares with intervening remissions | Good; variable disability |
| Chronic articular | 20-30% | Persistent arthritis with less prominent systemic features | Guarded; risk of joint destruction |
Prognostic Factors
Poor Prognostic Factors (associated with chronic articular pattern):
- Hip or shoulder involvement at onset
- Polyarticular disease (≥5 joints)
- Need for > 2 years of corticosteroids
- Thrombocytosis at presentation
- Age > 36 years at onset (some studies)
Good Prognostic Factors:
- Monocyclic pattern
- Prominent systemic features with less arthritis
- Younger age at onset
- Rapid response to treatment
Mortality
Overall mortality is low in modern era with effective therapies:
- Overall mortality: 3-5% in contemporary series (higher in older studies)
- Causes of death:
- MAS (most common preventable cause)
- Infection (especially with immunosuppression)
- Cardiac complications (tamponade, myocarditis)
- Amyloidosis (rare now)
- ARDS
- Mortality predictors: MAS, severe organ involvement, delayed diagnosis, infection
Functional Outcomes
- Monocyclic/polycyclic: Usually maintain good function
- Chronic articular: 20-30% develop significant disability
- Work disability: 15-20% in long-term follow-up
- Quality of life: Generally preserved with adequate treatment; chronic articular pattern has worse QOL
Remission Rates
With modern therapies (biologics):
- Complete remission: 40-60%
- Partial remission: 20-30%
- Refractory disease: 10-20%
Remission definition: Absence of clinical and laboratory features of disease activity off treatment
Long-Term Follow-Up
- Minimum 10 years recommended
- Monitor: Disease activity, treatment toxicity, amyloidosis, functional status
- Screening for amyloidosis: Urine protein, renal function annually
- Cardiovascular risk: Increased due to chronic inflammation and corticosteroid use
- Infection surveillance: Particularly on biologics
10. Special Populations
Pregnancy
Fertility: Not impaired by AOSD itself Disease activity in pregnancy: Variable; may improve, remain stable, or flare Maternal risks: Preterm delivery, preeclampsia, disease flare Fetal risks: Intrauterine growth restriction
Management:
- Conception planning: Optimize disease control before pregnancy
- Safe medications: Prednisolone (does not cross placenta well), azathioprine, hydroxychloroquine, anakinra (category B)
- Avoid: Methotrexate (teratogenic; discontinue 3 months before conception), NSAIDs (third trimester)
- Monitoring: Increased frequency; multidisciplinary care (rheumatology, obstetrics)
- Breastfeeding: Prednisolone and anakinra considered compatible; discuss individual medications
Elderly Patients
Presentation: May have less prominent systemic features; more organ involvement Differential: Infection and malignancy more common; require careful exclusion Treatment: Lower doses of medications; increased infection risk Comorbidities: Cardiovascular, renal disease common Prognosis: Worse than younger patients due to comorbidities and treatment complications
11. Key Guidelines and Consensus Statements
While there are no formal international guidelines specific to AOSD, several consensus statements and reviews guide management:
-
Gerfaud-Valentin et al. (2014) - Adult-onset Still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine. Comprehensive cohort study. [13]
-
Feist et al. (2018) - Mechanisms, biomarkers and targets for adult-onset Still's disease. Nature Reviews Rheumatology. Comprehensive pathophysiology review. [2]
-
Giacomelli et al. (2021) - A comprehensive review on adult onset Still's disease. Journal of Autoimmunity. Current management strategies. [1]
-
EULAR/ACR: No specific AOSD guidelines; general autoinflammatory disease principles apply
-
Japanese College of Rheumatology (JCR): Consensus statement on AOSD management (Japanese language)
12. Examination Focus
Common Exam Questions
1. "A 28-year-old woman presents with daily spiking fevers to 40°C for 2 weeks, sore throat, and a transient rash. How would you investigate?"
Model Answer: "This presentation raises concern for adult-onset Still's disease, though the differential is broad including infection, malignancy, and other rheumatic conditions. My approach would be systematic:
First, I would take a detailed history focusing on:
- Pattern of fever: quotidian pattern with evening spikes suggests AOSD
- Rash characteristics: salmon-pink, evanescent, appearing with fever
- Joint symptoms: arthralgia or arthritis
- Exclusion of infection sources and malignancy red flags
Examination would include:
- Observing the rash during a febrile episode (evanescent nature)
- Testing for dermographism
- Joint examination for synovitis
- Lymphadenopathy and hepatosplenomegaly
Investigations would include:
- Exclusion first: Blood cultures, viral serology (EBV, CMV, HIV), chest X-ray, tuberculin testing
- Diagnostic workup: CBC (leukocytosis with neutrophilia), ESR/CRP (markedly elevated), ferritin with glycosylated fraction (> 1000 with less than 20% glycosylation highly specific), LFTs, ANA/RF (negative in AOSD)
- If lymphadenopathy: CT chest/abdomen/pelvis; consider lymph node biopsy
- Apply Yamaguchi criteria: At least 5 criteria with at least 2 major, after exclusions
If AOSD confirmed, assess disease severity and consider treatment with corticosteroids, with early biologic therapy for moderate-severe disease."
2. "What is the role of ferritin in diagnosing AOSD?"
Model Answer: "Ferritin plays a crucial diagnostic role in AOSD. The key points are:
Markedly elevated ferritin: Typically > 1000 ng/mL, often > 3000-5000 ng/mL, and can exceed 50,000 ng/mL. A five-fold elevation above the upper limit of normal has 80% sensitivity for AOSD.
Glycosylated ferritin fraction: The critical discriminatory test. In AOSD, this is less than 20%, compared to the normal range of 50-80%. This has 70% sensitivity and 83% specificity for AOSD.
Pathophysiology: IL-6 and IL-1β drive ferritin production by hepatocytes and macrophages. In acute inflammation, the rapid production rate overwhelms the glycosylation machinery, resulting in a low glycosylated fraction. Ferritin also acts as a cytokine with immunomodulatory properties.
Differential diagnosis: Other causes of hyperferritinemia include hemochromatosis (normal glycosylated fraction), hemophagocytic syndromes (ferritin high but glycosylated fraction may be higher), liver disease, and malignancy. The combination of very high ferritin with low glycosylated fraction is relatively specific to AOSD.
Monitoring: Ferritin levels correlate with disease activity and can be used to monitor treatment response. However, paradoxically, falling ferritin with worsening clinical picture may indicate MAS, as the acute phase response fails."
3. "How do you recognize and manage macrophage activation syndrome in AOSD?"
Model Answer: "MAS is a life-threatening complication occurring in 10-15% of AOSD patients, with 10-20% mortality. Early recognition is critical.
Red flags for MAS:
- Change in fever pattern: continuous fever replacing the quotidian pattern
- Falling platelet count and other cytopenias
- Paradoxically falling ferritin (though still elevated) as acute phase response fails
- Rising liver enzymes and LDH
- Altered mental status
- Coagulopathy with falling fibrinogen, elevated D-dimer
Diagnostic criteria (HLH-2004):
- Fever
- Splenomegaly
- Cytopenias (≥2 lineages)
- Hypertriglyceridemia (> 265 mg/dL)
- Hypofibrinogenemia (less than 150 mg/dL)
- Hemophagocytosis on bone marrow
- Elevated soluble IL-2 receptor
- Low or absent NK cell activity
Management is urgent and aggressive:
- High-dose IV methylprednisolone: 1000 mg daily for 3-5 days
- Anakinra: 100 mg SC daily or higher doses (2-4 mg/kg/day). Rapid onset makes this ideal.
- Cyclosporine: 2-5 mg/kg/day, particularly effective for MAS
- Consider etoposide in refractory cases (HLH protocol)
- ICU support with transfusion support, treatment of coagulopathy
- Treat any triggering infection
Early recognition and aggressive treatment are essential to reduce mortality."
4. "What are the Yamaguchi criteria for AOSD?"
Model Answer: "The Yamaguchi criteria (1992) are the most widely used diagnostic classification for AOSD. Diagnosis requires at least 5 criteria including at least 2 major criteria, after exclusion of infections, malignancies, and other rheumatic diseases.
Major criteria (need ≥2):
- Fever ≥39°C lasting ≥1 week
- Arthralgia or arthritis lasting ≥2 weeks
- Typical rash: salmon-pink, evanescent, macular
- Leukocytosis ≥10,000/μL with ≥80% granulocytes
Minor criteria:
- Sore throat
- Lymphadenopathy and/or splenomegaly
- Liver enzyme abnormalities
- Negative RF and negative ANA
Sensitivity 96%, specificity 92% when applied correctly with appropriate exclusions.
The Fautrel criteria (2002) are an alternative that incorporates glycosylated ferritin ≤20% as a major criterion, with sensitivity 81% and specificity 98%."
5. "Compare IL-1 and IL-6 inhibitors in AOSD treatment."
Model Answer: "Both IL-1 and IL-6 inhibitors are highly effective in AOSD, but have different characteristics:
IL-1 inhibitors (Anakinra, Canakinumab):
- Mechanism: Block IL-1β signaling, central to AOSD pathogenesis
- Systemic symptoms: Very rapid response (24-48 hours for fever/rash)
- Articular symptoms: Good response but may be incomplete
- MAS: Effective with rapid onset
- Dosing: Anakinra 100 mg SC daily; Canakinumab 4 mg/kg monthly
- Advantages: Rapid onset, safe in pregnancy, short half-life for anakinra
- Limitations: Daily injection for anakinra, injection site reactions
IL-6 inhibitors (Tocilizumab, Sarilumab):
- Mechanism: Block IL-6 receptor, reduces inflammation and ferritin
- Systemic symptoms: Effective (1-2 weeks)
- Articular symptoms: Excellent, particularly for chronic articular disease
- Dosing: Tocilizumab 8 mg/kg IV monthly or 162 mg SC weekly
- Advantages: Excellent for chronic arthritis, less frequent dosing
- Limitations: Lipid abnormalities, masks CRP (complicates infection diagnosis), neutropenia risk
Choosing:
- Acute systemic disease or MAS: Anakinra first-line (rapid onset)
- Chronic articular pattern: Tocilizumab may be preferred
- Switch if inadequate response at 3 months
Both have response rates of 60-80% and are superior to traditional DMARDs."
Viva Points
Opening Statement: "Adult-onset Still's disease is a rare systemic autoinflammatory disorder characterized by quotidian fever, evanescent salmon-pink rash, and arthritis. It represents dysregulation of the innate immune system with overproduction of IL-1β, IL-6, and IL-18, distinguishing it from autoimmune diseases."
Key Facts to Mention:
- Incidence: 0.16-0.4 per 100,000; bimodal age distribution (15-25 and 36-46 years)
- Diagnosis: Yamaguchi criteria (≥5 criteria including ≥2 major) after exclusions
- Pathognomonic finding: Ferritin > 1000 ng/mL with glycosylated fraction less than 20%
- Diagnosis of exclusion: Must rule out infection, malignancy, other rheumatic disease
- Treatment: Corticosteroids first-line; biologics (IL-1 inhibitors like anakinra, IL-6 inhibitors like tocilizumab) for moderate-severe disease
- Complication: MAS in 10-15%, life-threatening
- Prognosis: Three patterns (monocyclic 30-40%, polycyclic 30-40%, chronic articular 20-30%)
Evidence to Cite:
- "Yamaguchi criteria have 96% sensitivity and 92% specificity"
- "Glycosylated ferritin less than 20% has 83% specificity for AOSD"
- "IL-1 inhibitors show 60-70% complete response rates with rapid onset"
- "MAS occurs in 10-15% with 10-20% mortality despite treatment"
Common Mistakes
❌ Mistakes That Fail Candidates:
-
Failing to exclude infection and malignancy: AOSD is a diagnosis of exclusion. Never diagnose without systematic exclusion of sepsis, endocarditis, tuberculosis, and lymphoma.
-
Missing MAS: Not recognizing the red flags (continuous fever, cytopenias, falling ferritin paradoxically, coagulopathy). MAS is life-threatening and requires urgent treatment.
-
Not ordering glycosylated ferritin: Ordering total ferritin alone without the glycosylated fraction misses the most specific test for AOSD.
-
Incorrect Yamaguchi criteria application: Forgetting that negative ANA/RF is a minor criterion, or not counting correctly (need ≥2 major criteria).
-
Outdated treatment: Recommending methotrexate or azathioprine as first-line for moderate-severe disease instead of biologics (IL-1/IL-6 inhibitors).
-
Ignoring disease pattern: Not recognizing that chronic articular pattern requires different management approach (IL-6 inhibitors preferred).
-
Wrong biologic choice: Not understanding when to use IL-1 vs IL-6 inhibitor (IL-1 for acute/systemic, IL-6 for chronic articular).
Key Terminology
- Quotidian fever: Once-daily fever spike (distinguishes from other patterns)
- Evanescent rash: Transient, appearing with fever then fading
- Autoinflammatory vs autoimmune: Innate vs adaptive immunity
- Yamaguchi criteria: Diagnostic classification
- Glycosylated ferritin fraction: The specific diagnostic marker
- Macrophage activation syndrome (MAS): Life-threatening complication
- Monocyclic/polycyclic/chronic articular: Three disease patterns
References
-
Giacomelli R, et al. A comprehensive review on adult onset Still's disease. J Autoimmun. 2018;93:24-36. doi:10.1016/j.jaut.2018.07.018
-
Feist E, et al. Mechanisms, biomarkers and targets for adult-onset Still's disease. Nat Rev Rheumatol. 2018;14(10):603-618. doi:10.1038/s41584-018-0081-x
-
Yamaguchi M, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19(3):424-430. PMID:1578458
-
Fautrel B, et al. Proposal for a new set of classification criteria for adult-onset Still disease. Medicine (Baltimore). 2002;81(3):194-200. doi:10.1097/00005792-200205000-00003
-
Gerfaud-Valentin M, et al. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014;93(2):91-99. doi:10.1097/MD.0000000000000021
-
Asanuma YF, et al. Nationwide epidemiological survey of 169 patients with adult Still's disease in Japan. Mod Rheumatol. 2015;25(3):393-400. doi:10.3109/14397595.2014.974881
-
Magadur-Joly G, et al. Epidemiology of adult Still's disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54(7):587-590. doi:10.1136/ard.54.7.587
-
Cagatay Y, et al. Adult-onset Still's disease. Int J Clin Pract. 2009;63(7):1050-1055. doi:10.1111/j.1742-1241.2007.01393.x
-
Ohta A, et al. Adult Still's disease: review of 228 cases from the literature. J Rheumatol. 1987;14(6):1139-1146. PMID:3325642
-
Joung CI, et al. Association between HLA-DR B1 and clinical features of adult onset Still's disease in Korea. Clin Exp Rheumatol. 2003;21(4):489-492. PMID:12942701
-
Efthimiou P, et al. Adult-onset Still's disease in focus: Clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum. 2021;51(4):858-874. doi:10.1016/j.semarthrit.2021.06.004
-
Wakai K, et al. Seasonal variation of disease onset in adult-onset Still's disease. Clin Rheumatol. 1997;16(4):375-379. doi:10.1007/BF02238944
-
Gerfaud-Valentin M, et al. Adult-onset Still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014;93(2):91-99. doi:10.1097/MD.0000000000000021
-
Rossi-Semerano L, et al. Tolerance and efficacy of off-label anti-interleukin-1 treatments in France: a nationwide survey. Orphanet J Rare Dis. 2015;10:19. doi:10.1186/s13023-015-0228-7
-
Chen DY, et al. Significant associations of proinflammatory cytokines with clinical and laboratory parameters in adult-onset Still's disease. Scand J Rheumatol. 2005;34(2):94-98. doi:10.1080/03009740410010317
-
Choi JH, et al. Serum cytokine profiles in patients with adult onset Still's disease. J Rheumatol. 2003;30(11):2422-2427. PMID:14677188
-
Kudela H, et al. Interleukin-18 (IL-18) in patients with adult-onset Still's disease. Clin Rheumatol. 2020;39(11):3443-3450. doi:10.1007/s10067-020-05068-0
-
Jamilloux Y, et al. Pathogenesis of adult-onset Still's disease: new insights from the juvenile counterpart. Immunol Res. 2015;61(1-2):53-62. doi:10.1007/s12026-014-8561-9
-
Kim HA, et al. Serum calprotectin levels in adult-onset Still's disease: clinical implications and comparison with procalcitonin levels. Scand J Rheumatol. 2015;44(4):297-303. doi:10.3109/03009742.2014.997427
-
Pouchot J, et al. Adult Still's disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991;70(2):118-136. doi:10.1097/00005792-199103000-00004
-
Nordström D, et al. Beneficial effect of interleukin 1 inhibition with anakinra in adult-onset Still's disease. An open, randomized, multicenter study. J Rheumatol. 2012;39(10):2008-2011. doi:10.3899/jrheum.111549
-
Kedor C, et al. Canakinumab for Treatment of Adult-Onset Still's Disease to Achieve Reduction of Arthritic Manifestation (CONSIDER): phase II, randomised, double-blind, placebo-controlled, multicentre, investigator-initiated trial. Ann Rheum Dis. 2020;79(8):1090-1097. doi:10.1136/annrheumdis-2020-217155
-
Ortiz-Sanjuán F, et al. Efficacy of tocilizumab in conventional treatment-refractory adult-onset Still's disease: multicenter retrospective open-label study of thirty-four patients. Arthritis Rheumatol. 2014;66(6):1659-1665. doi:10.1002/art.38398
-
Fautrel B, et al. Tumor necrosis factor alpha blocking agents in refractory adult Still's disease: an observational study of 20 cases. Ann Rheum Dis. 2005;64(2):262-266. doi:10.1136/ard.2004.024026
Last Updated: 2026-01-05
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Innate Immunity and Autoinflammatory Disease
Differentials
Competing diagnoses and look-alikes to compare.
- Systemic Lupus Erythematosus
- Sepsis and Infection Syndromes
- Hemophagocytic Lymphohistiocytosis
- Lymphoma
Consequences
Complications and downstream problems to keep in mind.
- Macrophage Activation Syndrome