Haemophagocytic Lymphohistiocytosis
Haemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome characterized by pathological... MRCP, USMLE Step 2/3 exam preparatio
What matters first
Haemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome characterized by pathological... MRCP, USMLE Step 2/3 exam preparatio
Ferritin greater than 10,000 μg/L (highly specific for HLH)
8 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Ferritin greater than 10,000 μg/L (highly specific for HLH)
- Progressive multi-organ failure despite broad-spectrum antibiotics
- Rapidly falling fibrinogen with rising transaminases
- Refractory fever with bi-/tri-lineage cytopenias
Exam focus
Current exam surfaces linked to this topic.
- MRCP
- USMLE Step 2/3
- ICU Medicine
Linked comparisons
Differentials and adjacent topics worth opening next.
- Septic Shock
- Adult-Onset Still's Disease
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Haemophagocytic Lymphohistiocytosis
1. Clinical Overview
Summary
Haemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome characterized by pathological immune activation with excessive cytokine release, leading to fever, cytopenias, hepatosplenomegaly, coagulopathy, and multi-organ dysfunction. The condition arises from failure of the immune system to appropriately terminate an inflammatory response, resulting in uncontrolled proliferation and activation of macrophages and cytotoxic T lymphocytes that infiltrate multiple organs. [1,2]
HLH exists in two major forms: primary (familial) HLH, caused by genetic defects in cytotoxic lymphocyte function, and secondary (acquired) HLH, triggered by infections, malignancies, or rheumatological diseases. In adults, secondary HLH predominates and carries mortality rates of 40-75% even with treatment, making rapid recognition and intervention critical. [3,4] When HLH occurs in the context of rheumatological disease, it is often termed macrophage activation syndrome (MAS).
The hallmark laboratory finding is markedly elevated serum ferritin, with levels exceeding 10,000 μg/L demonstrating 90% sensitivity and 96% specificity for HLH. [5] Diagnosis relies on the HLH-2004 criteria, requiring five of eight clinical and laboratory parameters. Treatment involves addressing the underlying trigger combined with immunosuppressive therapy, typically with dexamethasone and etoposide-based protocols adapted from the HLH-94/2004 trials. [1] For primary HLH, allogeneic hematopoietic stem cell transplantation represents the only curative option.
Key Facts
- Definition: Hyperinflammatory syndrome from dysregulated immune activation with uncontrolled macrophage and cytotoxic T-cell proliferation
- Incidence: 1-2 per million in general population; likely severely underdiagnosed in adults [6]
- Primary HLH: 1 in 50,000-100,000 live births; predominantly pediatric presentation
- Secondary HLH: Far more common in adults; median age 40-60 years at presentation
- Mortality: 40-75% overall; approaches 100% if untreated; primary HLH fatal without stem cell transplant [3,4]
- Common Triggers: Epstein-Barr virus (most common infectious), T-cell/NK-cell lymphoma (most common malignant), systemic lupus erythematosus/Still's disease (rheumatological)
- Pathognomonic Finding: Ferritin > 10,000 μg/L has 90% sensitivity and 96% specificity [5]
- Diagnostic Standard: HLH-2004 criteria (≥5 of 8 parameters) [1]
- First-line Treatment: Address underlying trigger + immunosuppression (dexamethasone ± etoposide) [2]
- Time-Critical: Median time from symptom onset to death without treatment: 6-8 weeks
- Prognosis: Varies by etiology - infection-triggered (30-50% mortality), malignancy-triggered (> 70% mortality), MAS with appropriate treatment (15-30% mortality)
Clinical Pearls
Diagnostic Pearl: A ferritin greater than 10,000 μg/L in the context of fever and cytopenias should trigger immediate consideration of HLH, even before all HLH-2004 criteria are fulfilled. This threshold provides excellent specificity and should prompt urgent specialist consultation. [5]
Treatment Pearl: Do not delay immunosuppression while awaiting complete diagnostic workup if clinical suspicion is high (HScore > 169 or ferritin > 10,000 with compatible presentation). Each day of delayed treatment increases mortality risk. Initiate dexamethasone 10 mg/m² daily while continuing diagnostic evaluation. [2]
Pitfall Warning: Hemophagocytosis on bone marrow biopsy is neither sensitive nor specific for HLH diagnosis. Only 60-80% of confirmed HLH cases demonstrate hemophagocytosis at presentation, and it can be seen in sepsis, malignancy, and transfusion reactions. Never delay treatment waiting for marrow evidence. [7]
Pitfall Warning: Adult HLH frequently mimics septic shock. However, lack of response to broad-spectrum antibiotics and pressors with rising ferritin (> 2,000-3,000) should immediately raise suspicion for HLH rather than prompting escalation of antimicrobials alone.
Mnemonic: HLH causes F.E.V.E.R. CHAOS
- Ferritin sky-high (> 10,000)
- EBV (and other viral triggers)
- Very low fibrinogen
- Elevated triglycerides
- Refractory fever
- Cytopenias (bi/tri-lineage)
- Hepatosplenomegaly
- Activated macrophages (hemophagocytosis)
- Organ failure (multi-system)
- Soluble IL-2 receptor (sCD25) elevated
Why This Matters Clinically
HLH represents one of the most commonly missed diagnoses in critically ill patients, with delays in recognition directly correlating with mortality. The syndrome crosses multiple specialty boundaries—presenting to hematology, rheumatology, oncology, infectious diseases, and intensive care—yet remains unfamiliar to many clinicians outside specialist centers. [6]
Early recognition (median time to diagnosis: 7-14 days) and prompt initiation of immunosuppression can reduce mortality from > 90% to 40-50% in secondary HLH. [3] The condition is increasingly recognized in adults, particularly in association with EBV infection, T-cell lymphomas, and immune checkpoint inhibitor therapy—all growing clinical scenarios. Understanding the diagnostic approach and management principles is now essential for acute medicine, hematology, and critical care practice.
2. Epidemiology
Incidence and Prevalence
Primary (Familial) HLH:
- Estimated incidence: 1 in 50,000-100,000 live births [8]
- Predominantly affects infants and young children (median onset: 2-6 months)
- Adult-onset primary HLH rare but increasingly recognized (10-15% of primary HLH cases) [9]
- Higher incidence in populations with consanguinity
- No gender predilection
Secondary (Acquired) HLH:
- Estimated incidence in adults: 0.4-1.2 per million per year (likely significant underascertainment) [6]
- True incidence unknown due to diagnostic underrecognition
- ICU studies suggest HLH may account for 1-4% of unexplained febrile illnesses with organ dysfunction [10]
- Male predominance in adult series (M:F ratio approximately 1.5-2:1)
- Median age at presentation: 40-60 years for infection/malignancy-associated; 20-40 years for rheumatological disease-associated (MAS)
Demographics
Age Distribution:
- Primary HLH: Bimodal—major peak in infancy (less than 2 years: 70-80%), minor peak in young adults with delayed-onset genetic forms
- Secondary HLH: Can occur at any age; peak incidence in 5th-6th decades
- MAS: Younger demographic (median 25-35 years) reflecting underlying rheumatological disease population
Geographic and Ethnic Variations:
- Primary HLH: Higher rates in regions with increased consanguinity (Middle East, parts of Asia)
- Specific genetic variants show ethnic clustering (e.g., PRF1 A91V in African populations)
- EBV-associated HLH: Particularly prevalent in East Asian populations [11]
- No consistent geographic pattern for secondary HLH
Risk Factors
For Secondary HLH Development:
| Risk Category | Specific Factors | Relative Risk |
|---|---|---|
| Immunodeficiency | HIV/AIDS (CD4 less than 200) | 20-50× |
| Primary immunodeficiency disorders | 10-100× | |
| Post-transplant (solid organ/HSCT) | 10-30× | |
| Malignancy | T-cell lymphoma | 15-25× |
| NK-cell lymphoma | 30-50× | |
| Acute leukemia during treatment | 5-10× | |
| Infection | EBV (primary or reactivation) | 10-30× |
| CMV in immunocompromised | 5-15× | |
| Disseminated tuberculosis | 3-8× | |
| HIV seroconversion | 5-10× | |
| Rheumatological | Systemic JIA/Adult-onset Still's | 10-20× for MAS |
| Systemic lupus erythematosus | 5-10× for MAS | |
| Kawasaki disease (children) | 2-5× | |
| Iatrogenic | Immune checkpoint inhibitors | 5-15× |
| CAR-T cell therapy | 10-25× | |
| Immunosuppressive therapy | 2-5× | |
| Genetic | Heterozygous perforin mutations | 3-7× |
| HLA-linked susceptibility | 2-4× |
Triggers for Secondary HLH
Infectious (50-60% of adult secondary HLH):
| Pathogen Category | Common Agents | Notes |
|---|---|---|
| Viral | EBV (40-50% of infection-associated) | Most common overall trigger |
| Cytomegalovirus (15-20%) | Especially in immunocompromised | |
| HIV (acute/advanced disease) | Both seroconversion and AIDS | |
| Herpes simplex virus | Disseminated infection | |
| Varicella-zoster virus | Severe/disseminated disease | |
| Parvovirus B19 | Particularly in immunocompromised | |
| Influenza A/B | Seasonal peaks; H1N1 association | |
| SARS-CoV-2 | Severe COVID-19; cytokine storm overlap | |
| Dengue, Chikungunya | Endemic regions | |
| Bacterial | Mycobacterium tuberculosis | Disseminated/miliary TB |
| Salmonella species | Particularly S. typhi | |
| Brucellosis | Endemic regions | |
| Rickettsia | Particularly scrub typhus | |
| Fungal | Histoplasma capsulatum | Disseminated disease |
| Candida species | Severe invasive candidiasis | |
| Cryptococcus | CNS/disseminated infection | |
| Parasitic | Leishmania species | Visceral leishmaniasis |
| Plasmodium species | Severe malaria |
Malignancy-Associated (20-30% of adult secondary HLH):
| Malignancy Type | Frequency | Key Features |
|---|---|---|
| T-cell lymphoma | 40-50% of malignancy-HLH | Peripheral, ALCL, PTCL-NOS |
| NK-cell lymphoma | 15-25% | Particularly aggressive |
| B-cell lymphoma | 10-15% | Less common; Hodgkin's rare |
| Acute leukemia | 10-15% | During induction/relapse |
| Myelodysplastic syndrome | 2-5% | Rare but recognized |
| Solid tumors | less than 5% | Very rare; case reports only |
Rheumatological Disease-Associated (MAS) (15-20% of adult secondary HLH):
- Systemic-onset juvenile idiopathic arthritis (sJIA): 10-30% develop MAS
- Adult-onset Still's disease: 10-15% develop MAS [12]
- Systemic lupus erythematosus: 1-5% develop MAS; higher in Asian populations
- Kawasaki disease (pediatric): 1-3% develop MAS
- Other: Dermatomyositis, systemic sclerosis, vasculitis (rare)
Drug-Induced (5-10% of adult secondary HLH):
- Immune checkpoint inhibitors (anti-PD-1, anti-PD-L1, anti-CTLA-4): Increasing recognition
- CAR-T cell therapy: Overlaps with cytokine release syndrome (CRS)
- Immunosuppressive agents: Paradoxical HLH triggering
- Anticonvulsants: Rare but reported (phenytoin, carbamazepine)
- Biologics: Anti-TNF agents (rare)
Seasonal and Temporal Patterns
- EBV-associated HLH: No strong seasonal pattern
- Influenza-associated HLH: Winter predominance in temperate climates
- Dengue-associated HLH: Correlates with monsoon season in endemic areas
- COVID-19 association: Mirrored pandemic waves (2020-2022)
3. Pathophysiology
Normal Immune Regulation (For Comparison)
Physiological Immune Response:
- Antigen Presentation: Antigen-presenting cells (APCs) activate T-cells via MHC complexes
- T-cell Activation: CD8+ cytotoxic T-lymphocytes (CTLs) and NK cells proliferate and produce IFN-γ
- Target Cell Elimination: CTLs/NK cells release cytotoxic granules (perforin, granzymes) to kill infected/abnormal cells
- Apoptosis Induction: Perforin creates pores; granzymes enter target cells and trigger apoptosis
- Immune Termination: Successful killing removes antigenic stimulus → CTL/NK cell contraction → resolution of inflammation
- Homeostasis Restored: Cytokine levels normalize; tissue repair begins
Pathological Mechanisms in HLH
Primary Defect: Failed Cytotoxic Function
In HLH, genetic (primary HLH) or acquired (secondary HLH) impairment of cytotoxic function prevents effective target cell killing, leading to persistent immune stimulation. [13]
Phase 1: Initiating Trigger and Impaired Cytotoxicity
Primary (Familial) HLH:
- Germline mutations in genes essential for cytotoxic granule trafficking, docking, priming, or pore formation
- Most common: PRF1 (perforin, 20-40%), UNC13D (Munc13-4, 15-25%), STX11 (syntaxin-11, 10-15%), STXBP2 (Munc18-2, 5-10%)
- Result: CTLs/NK cells cannot effectively kill target cells via granule exocytosis pathway
- Persistent antigenic stimulation from unchecked infected/abnormal cells
Secondary HLH:
- Overwhelming antigen load (e.g., high EBV viral burden, disseminated lymphoma)
- Acquired dysfunction of cytotoxic machinery (unclear mechanisms; possibly viral interference, immune exhaustion)
- Similar functional endpoint: Ineffective target cell clearance → persistent stimulation
Phase 2: Persistent Immune Activation
Failed Immune Contraction:
- Inability to eliminate target cells → continued antigen presentation
- Sustained activation of CD8+ T-cells and NK cells
- Continuous production of IFN-γ (primary driver of pathology)
- IFN-γ activates macrophages → M1 polarization → further cytokine production
Phase 3: Cytokine Storm
Hyperproduction of Pro-inflammatory Cytokines:
Activated T-cells and macrophages produce massive quantities of cytokines, creating a self-perpetuating inflammatory cascade. [14]
| Cytokine | Primary Source | Key Pathological Effects |
|---|---|---|
| IFN-γ | CD8+ T-cells, NK cells | Master regulator; macrophage activation; drives entire process |
| IL-1β | Activated macrophages | Fever, acute phase response, endothelial activation |
| IL-6 | Macrophages, T-cells | Hepatic acute phase protein synthesis (↑ ferritin, ↑ CRP); fever |
| IL-18 | Macrophages | IFN-γ production amplification; NK cell activation |
| TNF-α | Macrophages | Endothelial damage, capillary leak, hypotension; hepatotoxicity |
| IL-10 | T-cells, macrophages | Paradoxically elevated (failed regulatory attempt) |
| M-CSF, GM-CSF | T-cells | Macrophage proliferation and survival |
| sCD25 (sIL-2R) | Activated T-cells | Marker of T-cell activation; correlates with disease activity |
Key Mechanistic Insight: IFN-γ is the central driver. It activates macrophages, which produce IL-1, IL-6, IL-18, and TNF-α. IL-18 further drives IFN-γ production from T-cells and NK cells, creating a positive feedback loop that becomes autonomous and self-sustaining even if the original trigger resolves. [14]
Phase 4: Macrophage Activation and Proliferation
Excessive Macrophage Activity:
- IFN-γ induces macrophage proliferation and activation (hemophagocytic macrophages)
- Macrophages infiltrate multiple organs: Bone marrow, liver, spleen, lymph nodes, CNS
- Hemophagocytosis: Activated macrophages engulf erythrocytes, platelets, leukocytes, and their precursors
- Not specific to HLH (seen in sepsis, malignancy, transfusion)
- Not required for diagnosis
- Reflects severity of macrophage activation
Phase 5: Multi-Organ Damage
Hematological:
- Cytopenias: Direct hemophagocytosis + bone marrow infiltration + cytokine-mediated suppression
- "Anemia: Hemophagocytosis + chronic disease + marrow suppression"
- "Thrombocytopenia: Hemophagocytosis + consumption + impaired production"
- "Neutropenia: Hemophagocytosis + marrow infiltration (paradoxically, monocytes often preserved/elevated)"
- Coagulopathy:
- "Hypofibrinogenemia: Hepatic synthetic dysfunction + consumption"
- "Prolonged PT/aPTT: Factor consumption + hepatic failure"
- "Elevated D-dimer: DIC-like picture (though frank DIC less common than in sepsis)"
Hepatic:
- Macrophage infiltration of hepatic sinusoids and portal tracts
- Hepatocellular damage from TNF-α and other cytokines → ↑ transaminases
- Cholestatic injury → ↑ bilirubin
- Synthetic dysfunction → ↓ fibrinogen, ↓ albumin, ↑ PT
- Hyperferritinemia:
- Ferritin release from activated macrophages (direct)
- IL-1 and IL-6 stimulate ferritin synthesis (indirect)
- Levels > 10,000 μg/L highly specific (macrophage origin predominates) [5]
Splenic:
- Massive splenomegaly from macrophage and lymphocyte infiltration
- Red pulp expansion
- Splenic sequestration contributing to cytopenias
Metabolic:
- Hypertriglyceridemia: Cytokine-mediated suppression of lipoprotein lipase → impaired triglyceride clearance [15]
- Hyperglycemia: Cytokine effects + stress response
- Hypoalbuminemia: Hepatic dysfunction + capillary leak + catabolism
- Hyperuricemia: Cell turnover
Neurological:
- CNS involvement in 30-70% (often underrecognized) [16]
- Mechanisms:
- Perivascular lymphohistiocytic infiltration
- Cytokine-mediated neuronal damage
- Microvascular injury and hemorrhage
- Manifestations: Encephalopathy, seizures, ataxia, cranial neuropathies, myelopathy
Cardiovascular:
- Myocardial depression from cytokines (particularly TNF-α, IL-1)
- Capillary leak syndrome → distributive shock
- Hemodynamic instability mimicking septic shock
- Arrhythmias
Pulmonary:
- Acute respiratory distress syndrome (ARDS) from cytokine-mediated lung injury
- Pulmonary hemorrhage (rare but life-threatening)
- Infiltration of lung parenchyma
Renal:
- Acute kidney injury from hypotension, tubular injury (cytokines), and glomerular damage
- Hemophagocytic infiltration of kidneys (less common)
Phase 6: Vicious Cycle and Multi-Organ Failure
Without intervention, the cytokine storm becomes self-sustaining:
- Organ damage → tissue necrosis → further antigen release → more immune activation
- Hemodynamic instability → tissue hypoxia → more cellular damage → worsening inflammation
- Infection risk from immunosuppression (paradoxically, despite immune hyperactivation, patients have impaired antimicrobial responses due to immune exhaustion and functional defects)
- Progressive multi-organ failure → death (median survival without treatment: 6-8 weeks)
Genetic Basis of Primary HLH
Cytotoxic Granule Pathway Defects:
| Gene | Protein | Function | FHL Type | Inheritance | Frequency |
|---|---|---|---|---|---|
| PRF1 | Perforin | Pore formation in target cell membrane | FHL2 | AR | 20-40% |
| UNC13D | Munc13-4 | Priming of cytotoxic granules for fusion | FHL3 | AR | 15-25% |
| STX11 | Syntaxin-11 | SNARE protein for granule-membrane fusion | FHL4 | AR | 10-15% |
| STXBP2 | Munc18-2 | Regulates syntaxin-11 function | FHL5 | AR | 5-10% |
| RAB27A | Rab27a | Granule transport and docking | Griscelli syndrome type 2 | AR | less than 5% |
| LYST | Lysosomal trafficking regulator | Granule biogenesis/trafficking | Chédiak-Higashi syndrome | AR | less than 5% |
| AP3B1 | AP-3 complex | Vesicle trafficking | Hermansky-Pudlak syndrome type 2 | AR | less than 5% |
| SH2D1A | SAP (SLAM-associated protein) | NK/T-cell signaling | X-linked lymphoproliferative disease type 1 (XLP1) | XLR | 5-10% |
| BIRC4 | XIAP | Apoptosis regulation, NF-κB signaling | X-linked lymphoproliferative disease type 2 (XLP2) | XLR | less than 5% |
AR = Autosomal recessive; XLR = X-linked recessive
Genetic Testing Implications:
- Genetic confirmation identifies ~70-80% of primary HLH cases
- 20-30% of clinically diagnosed primary HLH have no identified mutation (likely novel genes or regulatory variants)
- Heterozygous carriers (e.g., single PRF1 mutation) have increased risk of secondary HLH under high antigen load [17]
- Genetic diagnosis critical for:
- Family counseling
- Sibling HLA typing for transplant
- Decision-making regarding stem cell transplant
- Prognostication
EBV as a Trigger: Special Considerations
EBV is the most common infectious trigger for HLH, particularly in East Asian populations. [11] Several unique features:
- Viral-Host Interaction: EBV infects and persists in B-lymphocytes; in susceptible individuals, failure to control EBV-infected B-cells leads to persistent antigen stimulation
- Chronic Active EBV (CAEBV): Distinct syndrome with clonal proliferation of EBV-infected T or NK cells; very high risk for HLH development
- EBV-HLH Phenotype: Often more aggressive, higher mortality, frequent CNS involvement
- Therapeutic Implications: May benefit from rituximab (anti-CD20) to reduce EBV-infected B-cell burden in addition to standard HLH therapy
MAS vs. HLH: Mechanistic Differences?
Macrophage activation syndrome (MAS) in rheumatological diseases shares the same final common pathway as HLH but may have distinct triggers:
- MAS Triggers: Disease flare, infection (concurrent), medication change
- Cytokine Profile: IL-1 and IL-18 particularly elevated (rationale for anakinra use) [18]
- Underlying Predisposition: Chronic immune activation from rheumatological disease may "prime" the system for MAS
- Milder Presentation: Some cases of MAS are less severe than malignancy/infection-associated HLH, though severe MAS is clinically indistinguishable
Despite these nuances, MAS and HLH represent a continuous spectrum of immune dysregulation rather than distinct entities.
4. Clinical Presentation
Timeline of Illness
Pre-diagnosis Phase (Days to Weeks):
- Initial Trigger Event: Viral illness, malignancy diagnosis/treatment, rheumatological flare
- Early Symptoms (often non-specific): Fever, malaise, fatigue, anorexia
- Subacute Evolution: Persistent fever despite antibiotics, worsening fatigue, development of organomegaly
- Critical Deterioration: Multi-organ dysfunction, shock, encephalopathy
- Median Time to Diagnosis: 7-14 days from symptom onset (often delayed)
- Median Time to Death (Untreated): 6-8 weeks from symptom onset
Post-diagnosis/Treatment Phase:
- Response to therapy typically seen within 7-14 days (falling ferritin, improving cytopenias)
- Refractory disease or early relapse: Poor prognostic sign
- Late complications: Infection, organ dysfunction, relapse (particularly if underlying trigger unresolved)
Symptoms
Constitutional (Nearly Universal):
- Fever: 90-97% of patients [1,2]
- High-grade (often > 39°C)
- Persistent/continuous (unlike intermittent fever in Still's disease, though overlap exists)
- Unresponsive to antibiotics and antipyretics
- May temporarily respond to corticosteroids
- Fatigue and Malaise: Severe, progressive
- Anorexia and Weight Loss: Rapid, significant
Gastrointestinal:
- Abdominal pain/discomfort: 30-40% (from hepatosplenomegaly)
- Nausea and vomiting: 20-30%
- Diarrhea: 10-20%
- Jaundice: 20-40% (hepatic involvement)
Neurological (30-70%, Often Underrecognized): [16]
- Encephalopathy: Confusion, altered mental status, lethargy (most common)
- Seizures: Focal or generalized
- Headache: Severe, persistent
- Ataxia and Dysmetria: Cerebellar involvement
- Cranial Neuropathies: Ophthalmoplegia, facial weakness
- Myelopathy: Weakness, sensory loss, bladder/bowel dysfunction
- Irritability (pediatric): Prominent early feature in children
Hemorrhagic:
- Easy bruising and petechiae: 20-40%
- Mucosal bleeding (epistaxis, gingival): 10-20%
- Serious hemorrhage (GI, pulmonary, CNS): 5-10% (severe cases)
Respiratory:
- Dyspnea: 20-40% (ARDS, pulmonary involvement)
- Cough: 10-20%
Dermatological:
- Rash: 20-40%
- Maculopapular eruption (most common)
- Petechial/purpuric (thrombocytopenia)
- Erythroderma (rare)
- Lymphedema (rare)
Signs
Vital Signs:
- Fever: Persistent high-grade (> 38.5°C, often > 39-40°C)
- Tachycardia: Proportionate to fever; may be disproportionate in shock
- Hypotension: Late sign; indicates decompensation/shock
- Tachypnea: ARDS, metabolic acidosis, or shock
General Examination:
- Pallor: Severe anemia
- Jaundice: Hepatic involvement (20-40%)
- Lymphedema: Rare but specific finding
Abdominal Examination:
- Hepatomegaly: 60-90% [1,2]
- Can be massive
- Tender
- Splenomegaly: 70-95% [1,2]
- Often marked (> 5 cm below costal margin)
- Cardinal feature; absence should raise doubt (though not absolute)
- Ascites: Uncommon unless severe hepatic dysfunction
Lymphatic:
- Lymphadenopathy: 30-60%
- Generalized or localized
- Variable size
- May indicate underlying malignancy (lymphoma)
Skin:
- Petechiae and Ecchymoses: Thrombocytopenia, coagulopathy
- Rash: Variable morphology (see above)
- Pallor: Anemia
Neurological Examination:
- Altered Mental Status: GCS reduction, confusion, delirium
- Focal Neurological Deficits: Hemiparesis, cranial nerve palsies, ataxia
- Meningism: Rare unless CNS infection co-exists
- Seizure Activity: Focal or generalized
Cardiovascular:
- Hypotension: Distributive shock (cytokine-mediated)
- Tachycardia: Compensatory or shock
- Peripheral edema: Capillary leak, hypoalbuminemia
Respiratory:
- Tachypnea: ARDS, metabolic compensation
- Crackles: Pulmonary edema, ARDS
- Hypoxemia: SpO₂ less than 92% on room air (severe cases)
Red Flags (Immediate Specialist Referral/HDU-ICU Admission)
[!CAUTION] Critical Red Flags Indicating Severe HLH with High Mortality Risk:
- Ferritin > 10,000 μg/L with compatible clinical picture (fever, cytopenias)
- Rapidly falling fibrinogen (less than 1.5 g/L or falling > 0.5 g/L per day) with rising transaminases
- Progressive multi-organ failure despite broad-spectrum antibiotics and resuscitation
- Refractory fever with bi- or tri-lineage cytopenias unresponsive to standard care
- New-onset neurological symptoms (confusion, seizures, focal deficits) with systemic inflammation
- Hemodynamic instability (shock) with hyperferritinemia and cytopenias
- Rapidly progressive hepatic dysfunction (transaminases > 1,000 IU/L, coagulopathy)
- HScore > 169 (> 90% probability of HLH) [19]
- Lactate dehydrogenase (LDH) > 1,000 IU/L with ferritin > 3,000 μg/L and cytopenias
Presentation Patterns by Etiology
Infection-Triggered HLH:
- Acute onset (days to 1-2 weeks)
- Clear infectious prodrome often identifiable
- High fever (> 39°C) universal
- Respiratory or GI symptoms may predominate initially
- Rapid progression
Malignancy-Associated HLH:
- Subacute onset (weeks)
- May have known malignancy (lymphoma diagnosis) or HLH as presenting feature
- B-symptoms often present (fever, night sweats, weight loss)
- Lymphadenopathy more prominent
- Slower progression but poorer prognosis
MAS (Rheumatological Disease-Associated HLH):
- Often in context of known rheumatological diagnosis (SLE, Still's disease)
- Acute deterioration during disease flare or after infection/medication change
- May have pre-existing organomegaly, cytopenias (need to recognize acute worsening)
- Rash more common (Still's: evanescent salmon-pink rash)
- Joint symptoms may be prominent (Still's)
Primary (Familial) HLH:
- Predominantly pediatric (infancy)
- Family history may be present (siblings, consanguinity)
- Recurrent or persistent presentation
- May have associated features (albinism in Griscelli, giant granules in Chédiak-Higashi)
- Adult-onset primary HLH: Consider in recurrent unexplained HLH or strong family history
Atypical Presentations
- Isolated CNS-HLH: Neurological symptoms without prominent systemic features (rare; requires high index of suspicion)
- Predominantly Hepatic: Mimics acute liver failure
- Renal-Predominant: AKI as presenting feature
- Post-Transplant: In setting of GVHD or infection; diagnostic confusion common
- Drug-Induced (Checkpoint Inhibitors): Overlaps with other immune-related adverse events (irAEs)
5. Investigations
Diagnostic Approach
HLH diagnosis is based on clinical and laboratory criteria. Two main frameworks exist:
- HLH-2004 Criteria: Standard diagnostic criteria (≥5 of 8 parameters) [1]
- HScore: Probability calculator for reactive HLH (score ≥169 suggests > 90% probability) [19]
HLH-2004 Diagnostic Criteria
Diagnosis of HLH Established by EITHER:
A. Molecular Diagnosis:
- Pathogenic mutation in genes associated with familial HLH (PRF1, UNC13D, STX11, STXBP2, RAB27A, LYST, AP3B1, SH2D1A, BIRC4)
OR
B. Fulfillment of 5 Out of 8 Clinical/Laboratory Criteria:
| Criterion | Threshold | Notes |
|---|---|---|
| 1. Fever | ≥38.5°C | Persistent or recurrent |
| 2. Splenomegaly | Palpable spleen | Clinical or radiological |
| 3. Cytopenias (≥2 lineages) | Hemoglobin less than 90 g/L (infants less than 100 g/L) Platelets less than 100 × 10⁹/L Neutrophils less than 1.0 × 10⁹/L | Not attributable to marrow hypoplasia |
| 4. Hypertriglyceridemia and/or Hypofibrinogenemia | Triglycerides ≥3.0 mmol/L (≥265 mg/dL) AND/OR Fibrinogen ≤1.5 g/L | Fasting triglycerides preferred |
| 5. Hemophagocytosis | Present in bone marrow, spleen, lymph node, or liver | No evidence of malignancy; not specific |
| 6. Low or Absent NK Cell Activity | less than 10% cytotoxicity at effector:target ratio of 50:1 | Not widely available; standardization issues |
| 7. Ferritin | ≥500 μg/L | > 10,000 μg/L highly specific (90% sens, 96% spec) [5] |
| 8. Soluble CD25 (sIL-2R) | ≥2,400 U/mL | Reflects T-cell activation; not widely available |
Limitations of HLH-2004:
- Developed primarily for pediatric familial HLH
- Some criteria (NK cell activity, sCD25) not widely available in routine practice
- Ferritin threshold of 500 μg/L too low (many conditions cause ferritin > 500)
- Does not weight criteria by specificity
- May miss early or atypical cases
HScore (Probability Calculator for Reactive HLH)
The HScore provides a probability estimate for HLH diagnosis and was developed specifically for adults with secondary HLH. [19]
HScore Variables and Points:
| Variable | Categories | Points |
|---|---|---|
| Known Immunosuppression | Yes | +18 |
| No | 0 | |
| Temperature (°C) | less than 38.4 | 0 |
| 38.4-39.4 | +33 | |
| > 39.4 | +49 | |
| Organomegaly | Hepatomegaly + Splenomegaly | +38 |
| Hepatomegaly OR Splenomegaly | +23 | |
| None | 0 | |
| Triglycerides (mmol/L) | less than 1.5 | 0 |
| 1.5-4.0 | +44 | |
| > 4.0 | +64 | |
| Ferritin (μg/L) | less than 2,000 | 0 |
| 2,000-6,000 | +35 | |
| > 6,000 | +50 | |
| AST (IU/L) | less than 30 | 0 |
| ≥30 | +19 | |
| Fibrinogen (g/L) | > 2.5 | 0 |
| ≤2.5 | +30 | |
| Cytopenias | 1 lineage | 0 |
| 2 lineages | +24 | |
| 3 lineages | +34 | |
| Hemophagocytosis on Marrow | Yes | +35 |
| No | 0 |
Maximum Score: 337
Interpretation:
- Score less than 90: Low probability of HLH (less than 1%)
- Score 90-168: Intermediate probability (need further workup)
- Score ≥169: High probability (> 90% probability of HLH; consider treatment) [19]
Advantages of HScore:
- Designed for adult secondary HLH
- Uses routinely available tests
- Provides probability rather than binary cutoff
- Validated in multicenter cohorts
Limitations:
- Developed in predominantly Caucasian population; may perform differently in other ethnicities
- Requires bone marrow to achieve highest scores (though not essential)
- Not validated for primary HLH or pediatric populations
Initial Laboratory Investigations
Essential Tests (All Patients with Suspected HLH):
| Test Category | Specific Tests | Expected Findings in HLH |
|---|---|---|
| Complete Blood Count | Hemoglobin Platelets WBC with differential | ↓ Hb (less than 90 g/L typical) ↓ Platelets (less than 100 typical, often less than 50) ↓ Neutrophils (less than 1.0 typical); ↑ or normal monocytes |
| Coagulation | PT/INR aPTT Fibrinogen D-dimer | ↑ PT/INR (hepatic dysfunction) ↑ aPTT (variable) ↓ Fibrinogen (less than 1.5 g/L in severe cases) ↑↑ D-dimer |
| Liver Function | AST, ALT Bilirubin (total, direct) Alkaline phosphatase Albumin | ↑↑ Transaminases (often > 500 IU/L; can exceed 1,000) ↑ Bilirubin (conjugated predominance) ↑ ALP (variable) ↓ Albumin |
| Renal Function | Creatinine, urea eGFR | Variable; ↑ in AKI (common in severe HLH) |
| Inflammatory Markers | CRP ESR Ferritin ⭐ | ↑ CRP (but often less elevated than expected for degree of inflammation) ↑ ESR ↑↑↑ Ferritin (> 10,000 μg/L in 50-70%; > 500 in ~95%) [5] |
| Lipid Profile | Triglycerides (fasting) | ↑ Triglycerides (> 3.0 mmol/L in ~60%) |
| Lactate Dehydrogenase | LDH | ↑↑ LDH (reflects cell turnover; often > 1,000 IU/L) |
HLH-Specific Investigations:
| Test | Method | Interpretation | Availability |
|---|---|---|---|
| Soluble CD25 (sIL-2R) | Serum ELISA | ≥2,400 U/mL supports HLH (part of HLH-2004 criteria) | Limited; specialist centers |
| NK Cell Activity | Flow cytometry-based cytotoxicity assay | less than 10% at 50:1 effector:target ratio supports HLH | Limited; specialist centers; poor standardization |
| Soluble CD163 | Serum ELISA | Elevated (> 6,500 ng/mL); marker of macrophage activation; not in formal criteria but increasingly used | Research/specialist centers |
Bone Marrow Examination
Indications:
- Confirm diagnosis (contributes to HLH-2004 and HScore)
- Exclude underlying malignancy (particularly lymphoma, leukemia)
- Assess hemophagocytosis
Findings:
| Feature | Description | Diagnostic Value |
|---|---|---|
| Hemophagocytosis | Macrophages engulfing erythrocytes, leukocytes, platelets, or precursors | Present in 60-80% at initial biopsy; neither sensitive nor specific [7] |
| Can repeat if initially negative and suspicion high | ||
| Hypocellularity | Reduced hematopoietic elements | Common (marrow suppression) |
| Lymphohistiocytic Infiltration | Increased activated macrophages and lymphocytes | Characteristic |
| Malignancy | Lymphoma cells, blasts | Critical to identify (changes management) |
| Dyserythropoiesis | Abnormal RBC precursors | Variable |
Critical Point: Absence of hemophagocytosis does NOT exclude HLH. Sensitivity increases with repeated sampling, but treatment should not be delayed awaiting marrow results if clinical suspicion is high.
Imaging
Indications:
- Assess organomegaly
- Exclude malignancy
- Evaluate CNS involvement (if neurological symptoms)
- Identify complications
Recommended Imaging:
| Modality | Indication | Typical Findings |
|---|---|---|
| Chest X-ray | Baseline; assess pulmonary involvement | Interstitial infiltrates (ARDS), lymphadenopathy |
| CT Chest/Abdomen/Pelvis | Staging; exclude lymphoma; assess organomegaly | Hepatosplenomegaly (universal), lymphadenopathy (30-60%), pulmonary infiltrates |
| MRI Brain | Neurological symptoms or signs | T2/FLAIR hyperintensities (white matter, basal ganglia, periventricular); leptomeningeal enhancement; atrophy (chronic) [16] |
| PET-CT | Suspected malignancy (lymphoma staging) | Hypermetabolic hepatosplenomegaly; lymphadenopathy; bone marrow uptake |
| Ultrasound Abdomen | Initial assessment if CT unavailable | Hepatosplenomegaly; ascites (if present) |
Microbiological and Virological Investigations
Essential Workup (Identify Infectious Triggers):
| Test | Purpose | Method |
|---|---|---|
| Blood Cultures | Bacterial/fungal sepsis | Aerobic/anaerobic bottles; fungal cultures if risk factors |
| EBV Serology | Acute EBV vs. reactivation | IgM VCA (acute), IgG VCA + EBNA (past); EBV PCR quantitative (most useful) |
| EBV PCR | Viral load | Plasma/whole blood; correlates with disease activity; high loads (> 10⁴-10⁵ copies/mL) significant |
| CMV PCR | Active CMV infection | Plasma; particularly in immunocompromised |
| HIV Serology | Underlying immunodeficiency; HIV-associated HLH | 4th generation Ag/Ab assay; confirm with Western blot or PCR if positive |
| Respiratory Viral Panel | Influenza, RSV, others | Nasopharyngeal swab PCR (seasonal context) |
| Hepatitis Serology | Hepatitis A, B, C | Particularly if transaminitis prominent |
| Parvovirus B19 PCR | Parvovirus-associated HLH | If aplastic crisis or rash |
| Mycobacterial Studies | Tuberculosis (disseminated) | Sputum/BAL AFB smear & culture; TB PCR; consider tissue biopsy |
| Fungal Studies | Histoplasma, Cryptococcus, others | Urine Histoplasma antigen (endemic areas); serum Cryptococcus antigen; cultures |
Additional Tests Based on Epidemiology/Exposure:
- Dengue, Chikungunya serology/PCR (tropical exposure)
- Leishmaniasis serology/PCR (Mediterranean, South Asia, Latin America)
- Rickettsial serology (travel, tick exposure)
- SARS-CoV-2 PCR (COVID-19 pandemic context)
Malignancy Workup
If Malignancy Suspected (lymphadenopathy, B-symptoms, atypical age, no clear infection):
| Investigation | Purpose |
|---|---|
| Lymph Node Biopsy | Diagnose lymphoma (excisional biopsy preferred over FNA) |
| Flow Cytometry | Blood/marrow; detect abnormal lymphocyte populations (T-cell, NK-cell clones) |
| Bone Marrow Biopsy | Marrow involvement by lymphoma/leukemia; hemophagocytosis |
| PET-CT | Lymphoma staging |
| Immunohistochemistry | Lymph node/marrow; T-cell markers (CD3, CD4, CD8), NK-cell markers (CD56), B-cell markers |
| T-cell Receptor (TCR) Gene Rearrangement | Clonality assessment (detect T-cell lymphoma) |
| EBV-Encoded RNA (EBER) In Situ Hybridization | Detect EBV in tumor cells (EBV+ T/NK lymphoma) |
Genetic Testing (Primary HLH Suspected)
Indications for Genetic Testing:
- Age less than 18 years at presentation (especially less than 2 years)
- Family history of HLH or unexplained infant deaths
- Consanguinity
- Recurrent or relapsing HLH
- Associated features (albinism, bleeding diathesis, neuropathy)
- Adult-onset HLH with no clear secondary trigger
- Planning hematopoietic stem cell transplant
Methods:
- Targeted gene panel: Sequence known HLH-associated genes (PRF1, UNC13D, STX11, STXBP2, etc.)
- Whole exome sequencing (WES): If panel negative but strong suspicion
- Functional assays: Perforin expression by flow cytometry; NK cell degranulation assay (CD107a)
Turnaround Time: Weeks to months (do not delay treatment)
Investigations to Monitor Disease Activity and Treatment Response
| Test | Frequency | Purpose |
|---|---|---|
| Ferritin | Every 2-3 days initially; weekly during treatment | Trending ferritin is best marker of response; falling levels indicate improvement [20] |
| CBC | Daily in acute phase; every 2-3 days during treatment | Monitor cytopenias, assess recovery |
| Fibrinogen | Every 2-3 days | Rising fibrinogen indicates improvement |
| Liver Enzymes | Twice weekly | Assess hepatic recovery; monitor etoposide toxicity |
| Triglycerides | Weekly | Should normalize with treatment |
| sCD25 (if available) | Weekly | Correlates with disease activity [21] |
Differential Diagnosis Considerations
HLH can mimic many conditions; key differentials and distinguishing features:
| Differential | Key Distinguishing Features | Overlapping Features |
|---|---|---|
| Septic Shock | Positive blood cultures; responds to antibiotics; ferritin rarely > 5,000 | Fever, cytopenias, shock, coagulopathy |
| Adult-Onset Still's Disease | Quotidian fever pattern; evanescent rash; arthralgias; no hemophagocytosis | Hyperferritinemia, hepatosplenomegaly (Still's can → MAS) |
| Lymphoma with B-symptoms | Lymphadenopathy; mass lesions; malignant cells on biopsy | Fever, cytopenias, LDH elevation (lymphoma can → HLH) |
| Acute Liver Failure | Hepatic encephalopathy; coagulopathy; transaminases often > 3,000-5,000 | Transaminitis, coagulopathy, low fibrinogen |
| Disseminated TB | Positive TB cultures/PCR; granulomas on biopsy; endemic exposure | Fever, cytopenias, hepatosplenomegaly (TB can → HLH) |
| Systemic Lupus (Flare) | ANA, dsDNA positive; complement consumption; specific organ involvement | Cytopenias, fever, multi-organ (SLE can → MAS) |
| Catastrophic Antiphospholipid Syndrome | Thrombosis (not cytopenias); positive aPL antibodies | Multi-organ failure, coagulopathy |
| Thrombotic Thrombocytopenic Purpura | Microangiopathic hemolysis (schistocytes); severe ADAMTS13 deficiency | Thrombocytopenia, fever, neuro symptoms |
Key Discriminator: Ferritin > 10,000 μg/L with fever and cytopenias strongly favors HLH over most differentials (exception: Still's disease, which can also have ferritin > 10,000 but usually has quotidian fever and different organ involvement pattern).
6. Management
Treatment Algorithm
Image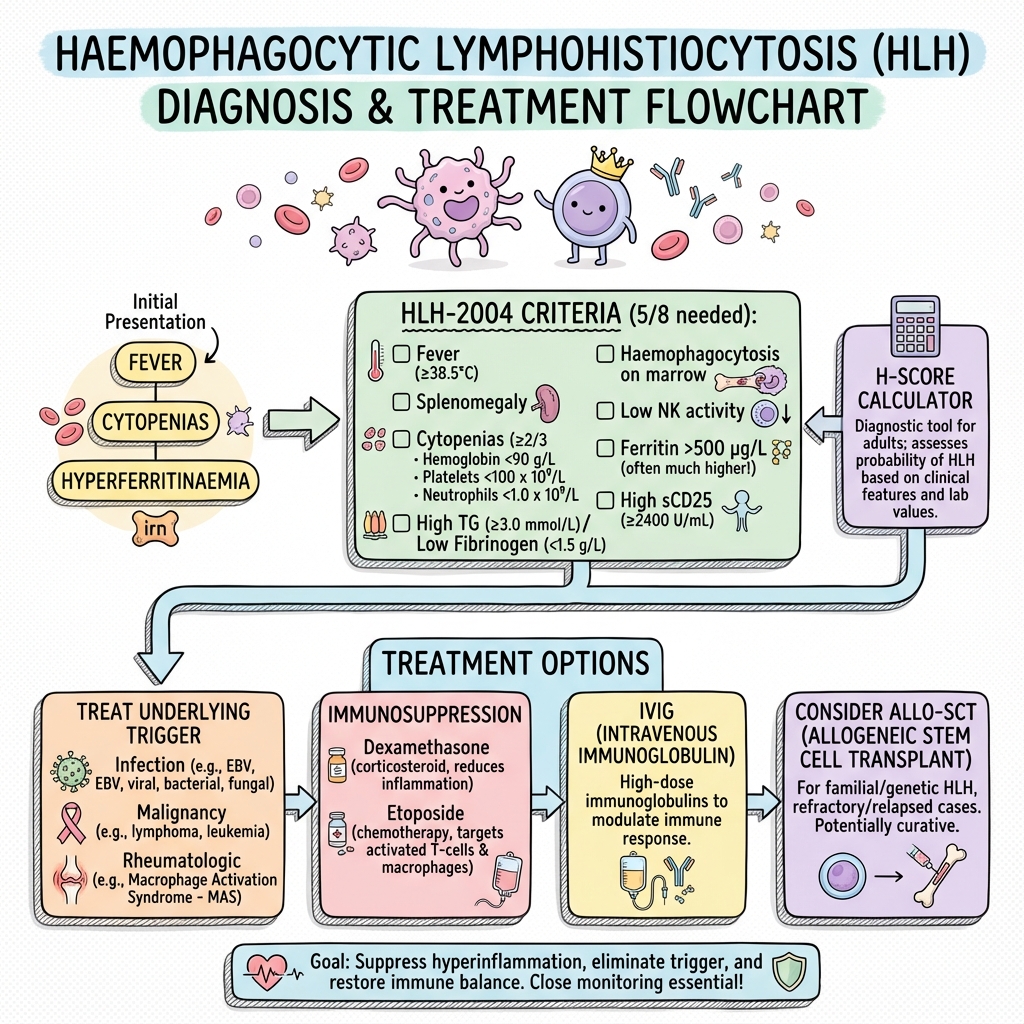 HLH Management Flowchart
HLH Management Flowchart
Key Principles:
- Treat Urgently: HLH is a medical emergency; delays increase mortality
- Dual Approach: (a) Treat underlying trigger AND (b) Suppress hyperinflammation
- Multidisciplinary Care: Involve hematology, ICU, infectious diseases, rheumatology as appropriate
- Supportive Care: Transfusions, organ support, infection prevention
- Definitive Therapy for Primary HLH: Allogeneic hematopoietic stem cell transplant (only curative option)
Initial Resuscitation and Supportive Care
Setting:
- High-dependency unit (HDU) or intensive care unit (ICU) for severe cases
- Specialist hematology ward with ICU backup for less severe cases
Immediate Actions:
| Intervention | Details |
|---|---|
| Hemodynamic Support | Fluid resuscitation (balanced crystalloids); vasopressors if shock (norepinephrine first-line) |
| Respiratory Support | Supplemental oxygen; non-invasive ventilation; mechanical ventilation if ARDS/respiratory failure |
| Transfusion Support | Platelets if less than 10-20 × 10⁹/L or bleeding; RBCs for Hb less than 70-80 g/L; FFP if active bleeding + coagulopathy |
| Infection Prophylaxis | Broad-spectrum antibiotics if febrile neutropenia or suspected infection; antifungal prophylaxis (fluconazole/micafungin); PJP prophylaxis (cotrimoxazole) if immunosuppression planned |
| Seizure Management | Benzodiazepines, antiepileptics as needed (levetiracetam preferred; avoid phenytoin due to drug interactions) |
| Nutrition | Enteral nutrition preferred; TPN if gut dysfunction |
| VTE Prophylaxis | LMWH if platelets > 30-50 × 10⁹/L and no active bleeding (balance thrombosis vs. hemorrhage risk) |
Treating the Underlying Trigger
Critical Step: Identify and address the inciting cause, as HLH may not fully resolve without treating the trigger.
Infection-Triggered HLH
| Pathogen | Treatment |
|---|---|
| EBV | No specific antiviral effective for EBV-HLH; rituximab (anti-CD20) may reduce EBV-infected B-cell burden (controversial; case series suggest benefit) [22]; standard HLH therapy primary treatment |
| CMV | Ganciclovir 5 mg/kg IV q12h OR foscarnet 60 mg/kg IV q8h (if ganciclovir-resistant or neutropenia) |
| HSV/VZV | Acyclovir 10 mg/kg IV q8h (HSV) or 10-15 mg/kg IV q8h (VZV) |
| HIV | Initiate antiretroviral therapy (ART); defer until HLH stabilized if very unwell (balance IRIS risk) |
| Influenza | Oseltamivir 75 mg PO bid (if within 48h of symptoms or severe disease) |
| Tuberculosis | Standard anti-TB therapy (rifampicin, isoniazid, pyrazinamide, ethambutol); consider adjunctive corticosteroids (in HLH protocol) |
| Fungal (Histoplasma, etc.) | Liposomal amphotericin B 3-5 mg/kg/day; transition to itraconazole for step-down |
| Bacterial Sepsis | Broad-spectrum antibiotics per local guidelines (e.g., piperacillin-tazobactam + vancomycin); tailor based on cultures |
Special Consideration - COVID-19-Associated HLH:
- Overlaps with cytokine storm syndrome
- Supportive care + corticosteroids (dexamethasone 6 mg daily per COVID protocols)
- Consider IL-6 inhibition (tocilizumab) if available
- Etoposide generally avoided unless clear HLH by HLH-2004 criteria (risk of immunosuppression in active viral infection)
Malignancy-Associated HLH
Principle: Treat underlying malignancy definitively; HLH will not resolve without malignancy control.
| Malignancy | Treatment Approach |
|---|---|
| T-cell/NK-cell Lymphoma | Lymphoma-specific chemotherapy (e.g., CHOP, CHOEP) + HLH-directed therapy (etoposide often overlaps); high-dose methotrexate if CNS involvement; consider auto-SCT or allo-SCT |
| B-cell Lymphoma | Standard lymphoma therapy (R-CHOP for DLBCL); rituximab useful; HLH-directed therapy as bridge |
| Acute Leukemia | Induction chemotherapy per leukemia subtype; HLH often improves with leukemia treatment |
| Hodgkin Lymphoma | ABVD or escalated BEACOPP; HLH rare but reported |
Challenge: Distinguishing malignancy-triggered HLH from lymphoma with hemophagocytosis (secondary phenomenon) can be difficult; both require lymphoma treatment.
Rheumatological Disease-Associated HLH (MAS)
Underlying Disease Management:
| Disease | Specific Treatment |
|---|---|
| Adult-Onset Still's Disease | High-dose corticosteroids + anakinra (IL-1 receptor antagonist); consider tocilizumab (IL-6 inhibitor) [18] |
| Systemic Lupus Erythematosus | Pulse methylprednisolone + cyclophosphamide or rituximab; address SLE flare |
| Systemic JIA | Anakinra; corticosteroids; avoid NSAIDs during MAS |
| Kawasaki Disease | IVIG 2 g/kg + aspirin; corticosteroids if refractory |
Anakinra in MAS:
- Dosing: 1-2 mg/kg/day SC (pediatric); 100 mg SC daily or bid (adult); can escalate to 4-8 mg/kg/day in severe cases
- Rapid onset of action (hours to days)
- Evidence: Case series and observational studies show efficacy; no RCTs [18]
- Well-tolerated; infection risk
Immunosuppressive Therapy for HLH
HLH-94/2004 Protocol (Standard of Care)
The HLH-94 and HLH-2004 trials established etoposide-based therapy as standard for HLH. [1] Originally developed for pediatric primary HLH, these protocols are adapted for adults with secondary HLH.
HLH-2004 Protocol Overview:
| Phase | Duration | Treatment |
|---|---|---|
| Initial (Induction) | 8 weeks | Etoposide + Dexamethasone (+ Ciclosporin from week 3*) |
| Continuation | Until SCT or week 32 | Dexamethasone pulses + Ciclosporin + Etoposide (reduced frequency) |
| CNS Therapy | If CNS involvement | Intrathecal methotrexate + hydrocortisone |
*Ciclosporin timing varies; some protocols start from week 1.
Detailed HLH-2004 Induction Regimen:
| Drug | Dose | Schedule | Duration |
|---|---|---|---|
| Dexamethasone | 10 mg/m²/day | Days 1-14 | Then taper: 5 mg/m² days 15-28; 2.5 mg/m² days 29-42; 1.25 mg/m² days 43-56 |
| Etoposide | 150 mg/m² IV | Twice weekly weeks 1-2 (days 1, 4, 8, 11, 15); then weekly weeks 3-8 | 8 weeks |
| Ciclosporin A | 6 mg/kg/day PO (divided bid) | Start week 3* (or week 1 in some protocols) | Continue; target trough 200-400 ng/mL |
Intrathecal Therapy (if CNS involvement at diagnosis or develops during treatment):
- Methotrexate: Age-adjusted dose (12 mg for adults) IT weekly until CSF clears, then q2 weeks
- Hydrocortisone: 20-25 mg IT with methotrexate
Adult-Modified Approaches
Rationale for Modification:
- Secondary HLH in adults often less aggressive than primary HLH in children
- Etoposide carries significant toxicity (myelosuppression, infection risk, secondary malignancy)
- Tailoring intensity to severity may reduce toxicity
Common Modifications:
| Scenario | Modification |
|---|---|
| Mild-Moderate HLH (ferritin 2,000-10,000; stable organs; no shock) | Dexamethasone alone initially; add etoposide if no response in 3-7 days OR use reduced-dose etoposide (100 mg/m² or 75 mg/m² weekly) |
| Severe HLH (ferritin > 10,000; shock; multi-organ failure) | Full-dose etoposide + dexamethasone from day 1 |
| MAS (Rheumatological) | Methylprednisolone 1 g IV daily × 3 days, then dexamethasone + anakinra; reserve etoposide for refractory cases [12] |
| Infection-Triggered (Controlled Infection) | Standard protocol; treat infection concurrently |
| Malignancy-Associated | Lymphoma chemotherapy (which often includes etoposide-like agents) may suffice; coordinate with oncology |
Dexamethasone vs. Methylprednisolone:
- Dexamethasone: Better CNS penetration (important given 30-70% CNS involvement); preferred in HLH-2004 protocol
- Methylprednisolone: Often used as pulse therapy in MAS (1 g IV daily × 3)
Alternative and Emerging Immunosuppressive Agents
For Refractory or Relapsed HLH:
| Agent | Mechanism | Dosing | Evidence | Comments |
|---|---|---|---|---|
| Anakinra | IL-1 receptor antagonist | 1-10 mg/kg/day SC (up to 800 mg/day reported) | Case series; effective in MAS and refractory HLH [18] | Rapid action; well-tolerated; first-line for MAS |
| Tocilizumab | IL-6 receptor antibody | 8 mg/kg IV (max 800 mg) q2-4 weeks | Case reports; used in MAS and COVID-HLH | Consider if IL-6 markedly elevated |
| Ruxolitinib | JAK1/2 inhibitor | 10-20 mg PO bid | Case series; promising in refractory HLH [23] | Blocks IFN-γ and cytokine signaling; oral agent; myelosuppression risk |
| Emapalumab | Anti-IFN-γ monoclonal antibody | 1 mg/kg IV twice weekly; escalate to 3-10 mg/kg based on response | RCT in primary HLH; FDA/EMA approved for refractory primary HLH [24] | Very expensive; specialist use; requires infection screening |
| Alemtuzumab | Anti-CD52 (T-cell/monocyte depletion) | 10-30 mg IV daily × 5-10 days | Case series; salvage therapy | Severe immunosuppression; infection risk |
| ATG (Anti-thymocyte Globulin) | T-cell depletion | 2.5-3.5 mg/kg IV daily × 3-5 days | Case reports; salvage | Severe immunosuppression |
| Rituximab | Anti-CD20 (B-cell depletion) | 375 mg/m² IV weekly × 4 | Case series in EBV-HLH [22] | Targets EBV-infected B-cells; adjunct to HLH therapy |
| Infliximab | Anti-TNF-α | 5 mg/kg IV at 0, 2, 6 weeks | Limited data; case reports | Rarely used; infection risk |
Emapalumab (Gamifant®):
- First and only FDA-approved agent specifically for HLH (2018; primary refractory/relapsed HLH)
- Neutralizes IFN-γ (central cytokine in HLH pathophysiology)
- Evidence: Phase 2/3 trial (n=27) showed 63% overall response rate [24]
- Dosing: Start 1 mg/kg IV twice weekly; titrate based on response (max 10 mg/kg)
- Very costly (>$600,000 per treatment course in US)
- Requires extensive infection screening (TB, fungal, viral) before and during treatment
Ruxolitinib:
- JAK1/2 inhibitor; blocks signaling of IFN-γ, IL-6, and other cytokines
- Case series show promise in refractory HLH [23]
- Oral agent (advantage over IV therapies)
- Dosing: 10 mg PO bid initially; can escalate to 20 mg bid
- Toxicities: Myelosuppression, infection risk, GI upset
- Not yet standard of care; often used in trials or compassionate access
Hematopoietic Stem Cell Transplantation (HSCT)
Indications:
| Scenario | Recommendation |
|---|---|
| Primary (Familial) HLH | Allogeneic HSCT mandatory (only curative option); perform once disease controlled with chemotherapy |
| Refractory Secondary HLH | Consider allo-HSCT if no response to medical therapy and no other options |
| Relapsed Secondary HLH | Consider allo-HSCT, especially if multiple relapses |
| EBV-Driven HLH (Persistent) | Allo-HSCT may be considered if refractory to medical therapy |
| Malignancy-Associated HLH | Treat underlying malignancy; allo-HSCT per malignancy indication (e.g., relapsed lymphoma), not HLH per se |
Timing:
- Achieve disease control (falling ferritin, resolution of fever, improving cytopenias) before proceeding to transplant
- Typically after 8-12 weeks of HLH-2004 protocol
- Urgent transplant in uncontrolled disease carries prohibitive mortality
Donor Selection:
- Matched sibling donor (MSD) preferred
- Matched unrelated donor (MUD) acceptable
- Haploidentical or cord blood if no matched donor (higher risk)
Conditioning:
- Reduced-intensity conditioning (RIC) often used (lower toxicity)
- Myeloablative conditioning if young, fit patient
Outcomes:
- Primary HLH: 5-year survival post-HSCT 50-70% (vs. 0% without HSCT) [25]
- Refractory secondary HLH: Limited data; salvage option with lower success rates
Management of Specific Complications
CNS Involvement
Recognition:
- New neurological symptoms (seizures, confusion, focal deficits)
- MRI brain: T2/FLAIR hyperintensities, leptomeningeal enhancement
- CSF: Lymphocytic pleocytosis, elevated protein, normal/low glucose
Treatment:
- Dexamethasone: Better CNS penetration than other corticosteroids
- Intrathecal Therapy: Methotrexate + hydrocortisone weekly (see protocol above)
- Etoposide: Systemic etoposide has some CNS penetration
- Refractory: Consider emapalumab, allo-HSCT
Severe Coagulopathy/Bleeding
Management:
- Platelet Transfusion: Maintain platelets > 20-50 × 10⁹/L (higher if bleeding)
- FFP: If active bleeding and prolonged PT/aPTT
- Cryoprecipitate: If fibrinogen less than 1.0 g/L and bleeding
- Fibrinogen Concentrate: Alternative to cryoprecipitate (if available)
- Tranexamic Acid: Consider for mucosal bleeding (caution if DIC)
- Avoid Invasive Procedures unless absolutely necessary
Shock
Approach:
- Fluid resuscitation: 20-30 mL/kg boluses (balanced crystalloids)
- Vasopressors: Norepinephrine first-line
- Inotropes: Dobutamine or epinephrine if myocardial dysfunction
- Corticosteroids: Dexamethasone (as part of HLH therapy) provides some hemodynamic benefit
- Source Control: Treat infection if present
- Consider Early Immunosuppression: Cytokine storm is cause of shock; dexamethasone ± etoposide addresses pathophysiology
Infection During Immunosuppression
Prophylaxis:
- Antibacterial: Fluoroquinolone (levofloxacin) if prolonged neutropenia
- Antifungal: Fluconazole 200-400 mg daily OR micafungin/anidulafungin if high risk
- PJP: Cotrimoxazole DS three times weekly (or daily if severe immunosuppression)
- Antiviral: Acyclovir 400 mg bid (HSV prophylaxis); valganciclovir if CMV-seropositive and high risk
Management of Febrile Neutropenia:
- Broad-spectrum antibiotics (piperacillin-tazobactam or meropenem + vancomycin)
- Escalate to antifungals if persistent fever > 3-5 days
- G-CSF (filgrastim) if severe neutropenia, though benefit in HLH uncertain
Monitoring During Treatment
Clinical Monitoring:
- Daily assessment: Fever curve, organ function, neurological status
- HDU/ICU monitoring if severe
Laboratory Monitoring:
| Test | Frequency | Purpose |
|---|---|---|
| Ferritin | Every 2-3 days | Best marker of response; should fall progressively [20] |
| CBC | Daily initially; then every 2-3 days | Cytopenias should improve; monitor etoposide-induced myelosuppression |
| Fibrinogen, Triglycerides | Twice weekly | Should normalize |
| Liver Enzymes, Bilirubin | Twice weekly | Monitor hepatic recovery; etoposide hepatotoxicity |
| Creatinine | Twice weekly | Ciclosporin nephrotoxicity |
| Ciclosporin Trough | Twice weekly (once stable) | Target 200-400 ng/mL |
Response Criteria:
- Complete Response: Resolution of fever, normalization of ferritin and cytopenias, no organomegaly
- Partial Response: Improvement in 2+ parameters (e.g., falling ferritin, fever resolution, improving counts)
- No Response: Stable or worsening parameters after 2 weeks of therapy
- Relapse: Re-emergence of HLH features after initial response
Timeframe for Response:
- Fever: Should resolve within 7-10 days
- Ferritin: Should begin falling within 7 days; halving of peak ferritin by day 14-21 indicates response [20]
- Cytopenias: Slower recovery (2-4 weeks); platelets often recover first
Management Algorithm Summary
Suspected HLH (Fever + Cytopenias + Ferritin > 2,000-3,000)
↓
Initial Workup: HLH-2004 criteria, HScore, trigger workup (infection, malignancy, autoimmune)
↓
HLH Diagnosis Confirmed (≥5/8 HLH-2004 OR HScore ≥169)
↓
├─→ Identify and Treat Underlying Trigger (Infection, Malignancy, Rheumatological Disease)
↓
├─→ Assess Severity:
├─→ SEVERE (Ferritin > 10,000, shock, MOF): Etoposide 150 mg/m² + Dexamethasone 10 mg/m² (HLH-2004)
├─→ MODERATE (Ferritin 3,000-10,000, stable): Dexamethasone alone OR reduced etoposide
├─→ MAS (Rheumatological): Pulse methylprednisolone + Anakinra (reserve etoposide for refractory)
↓
Monitor Response (Ferritin, Cytopenias, Fever, Fibrinogen) at 7-14 days
↓
├─→ RESPONSE: Continue protocol; add ciclosporin week 3; taper dexamethasone per protocol
├─→ NO RESPONSE/REFRACTORY: Escalate (full HLH-2004 if not already; add anakinra, ruxolitinib, or emapalumab)
├─→ PRIMARY HLH: Plan allogeneic HSCT once disease controlled
↓
Continue Until:
- Complete response (ferritin less than 500, counts normalized, no fever) × 2-3 weeks → taper therapy
- Primary HLH → proceed to HSCT
- Refractory → salvage therapy or HSCT
7. Complications
Acute Complications (During Active Disease)
| Complication | Incidence | Clinical Features | Management |
|---|---|---|---|
| Multi-Organ Dysfunction Syndrome (MODS) | 50-70% | Simultaneous failure of liver, kidneys, lungs, coagulation | ICU support; organ-specific interventions; treat HLH aggressively |
| Disseminated Intravascular Coagulation (DIC) | 20-40% | Hypofibrinogenemia, elevated D-dimer, bleeding, thrombosis | FFP, platelets, cryoprecipitate; treat underlying HLH |
| Acute Respiratory Distress Syndrome (ARDS) | 20-30% | Bilateral infiltrates, hypoxemia, low compliance | Mechanical ventilation (lung-protective strategies); treat HLH |
| Shock (Distributive) | 30-50% in severe cases | Hypotension, tachycardia, end-organ hypoperfusion | Fluids, vasopressors, inotropes; corticosteroids (dexamethasone); treat HLH |
| Acute Kidney Injury | 30-40% | Rising creatinine, oliguria | Renal replacement therapy if severe; treat HLH; avoid nephrotoxins |
| CNS Involvement | 30-70% [16] | Seizures, encephalopathy, focal deficits, hemorrhage | Intrathecal methotrexate, dexamethasone, antiepileptics; MRI brain; treat HLH |
| Severe Bleeding | 10-20% | GI, pulmonary, CNS hemorrhage | Platelet/FFP transfusion; local hemostasis; treat coagulopathy |
| Severe Infection | 40-60% | Bacteremia, fungemia, viral reactivation | Broad-spectrum antimicrobials; prophylaxis; monitor closely |
| Hepatic Failure | 10-20% | Encephalopathy, severe coagulopathy, transaminases > 5,000 | Supportive; consider N-acetylcysteine; treat HLH; rarely requires liver transplant |
| Cardiac Dysfunction | 15-25% | Myocardial depression, arrhythmias, pericardial effusion | Inotropes, echocardiography monitoring, treat HLH |
Treatment-Related Complications
| Complication | Causative Agent | Incidence | Management |
|---|---|---|---|
| Severe Myelosuppression | Etoposide, ciclosporin | 30-50% | Dose reduction; G-CSF (benefit unclear); transfusion support; infection prophylaxis |
| Opportunistic Infections | Immunosuppression (etoposide, dexamethasone, ciclosporin) | 20-40% | Prophylaxis (PJP, fungal, viral); empiric treatment if febrile; monitor CMV, EBV, fungal markers |
| Secondary Malignancy | Etoposide | less than 5% (long-term risk) | Monitor; unavoidable given need for treatment |
| Hepatotoxicity | Etoposide, ciclosporin | 15-30% | Monitor LFTs; dose adjustment/discontinuation if severe |
| Nephrotoxicity | Ciclosporin | 10-20% | Monitor creatinine; dose adjustment; maintain trough 200-400 ng/mL |
| Hypertension | Ciclosporin, corticosteroids | 20-30% | Antihypertensives (avoid ciclosporin-interacting agents) |
| Hyperglycemia | Dexamethasone | 30-50% | Insulin therapy; monitor glucose |
| Posterior Reversible Encephalopathy Syndrome (PRES) | Ciclosporin, hypertension | 2-5% | MRI brain; discontinue ciclosporin; BP control; usually reversible |
| Avascular Necrosis | Corticosteroids | 2-5% (long-term) | Orthopedic referral; pain management; monitor |
| Osteoporosis | Corticosteroids | Variable (long-term) | Calcium, vitamin D, bisphosphonates; DEXA scan |
Long-Term Complications (Survivors)
| Complication | Incidence | Notes |
|---|---|---|
| Relapse of HLH | 10-30% (secondary HLH); 30-50% (primary HLH without HSCT) | Higher if underlying trigger unresolved (e.g., persistent EBV, uncontrolled lymphoma, genetic predisposition) |
| Chronic Organ Dysfunction | 10-20% | Renal impairment, hepatic fibrosis, neurological sequelae (cognitive, motor deficits) |
| Neurocognitive Impairment | 20-40% in CNS-HLH | Learning difficulties, memory problems, behavioral issues (particularly pediatric) |
| Chronic Fatigue | 30-50% | Multifactorial; often improves over months to years |
| Immunodeficiency | Variable | Particularly post-HSCT; increased infection risk |
| Secondary Malignancy | 2-5% at 10 years | Etoposide-associated (AML/MDS); HSCT-related (PTLD, solid tumors) |
| Endocrine Dysfunction | 10-20% | Hypothyroidism, growth hormone deficiency (pediatric), hypogonadism (post-HSCT) |
Mortality
Overall Mortality:
- Untreated HLH: > 95% mortality
- Treated Secondary HLH: 40-60% mortality [3,4]
- Treated Primary HLH without HSCT: 70-90% mortality
- Treated Primary HLH with HSCT: 30-50% mortality [25]
Mortality by Etiology:
- Infection-triggered (isolated, controlled infection): 30-50%
- Malignancy-associated HLH: 60-80% (driven by malignancy prognosis)
- MAS (rheumatological disease-associated): 10-30% (if treated appropriately)
- EBV-driven HLH: 50-70%
- Primary HLH (with HSCT): 30-50%
- Primary HLH (without HSCT): > 90%
Causes of Death:
- Multi-organ failure (40-50%)
- Sepsis/infection (30-40%)
- Hemorrhage (CNS, pulmonary, GI) (10-15%)
- Underlying malignancy progression (malignancy-HLH) (20-30%)
- Complications of HSCT (10-20% of transplanted patients)
8. Prognosis
Survival Outcomes
Primary HLH:
- Without Treatment: Fatal within weeks to months
- With HLH-2004 Protocol Alone: 50-60% initial response; 70-90% mortality long-term without HSCT
- With HLH-2004 Protocol + HSCT: 5-year overall survival 50-70% [25]
Secondary HLH:
- Overall: 40-60% mortality despite treatment [3,4]
- Infection-Triggered (Controlled): 30-50% mortality
- Malignancy-Associated: 60-80% mortality (largely driven by underlying malignancy)
- MAS: 10-30% mortality (better prognosis if treated appropriately; 5-10% in mild cases)
Prognostic Factors
Factors Associated with POOR Prognosis:
| Factor | Relative Risk of Death | Evidence |
|---|---|---|
| Delay in Diagnosis/Treatment | 2-3× | Each week delay increases mortality [3] |
| Age > 60 years | 1.5-2× | Poorer tolerance of treatment; comorbidities |
| Malignancy-Associated HLH | 2-3× | Underlying malignancy difficult to control |
| EBV-Driven HLH | 1.5-2.5× | High viral loads; aggressive course |
| CNS Involvement | 2-3× | Difficult to treat; indicates severe disease [16] |
| Ferritin > 50,000-100,000 μg/L | 2× | Extremely high ferritin indicates severe inflammation |
| Severe Multi-Organ Failure (≥3 organs) | 3-5× | Reflects advanced disease |
| Shock Requiring Vasopressors | 2-3× | Hemodynamic instability poor sign |
| No Response to Initial Therapy (by day 14) | 3-4× | Refractory disease portends poor outcome |
| Thrombocytopenia less than 20 × 10⁹/L | 1.5-2× | Severity marker |
| Hypofibrinogenemia less than 1.0 g/L | 2× | Coagulopathy severity |
| Elevated Lactate (> 4 mmol/L) | 2× | Tissue hypoxia, shock |
| Underlying Immunosuppression (HIV, transplant) | 1.5-2.5× | Impaired ability to control triggers |
Factors Associated with BETTER Prognosis:
| Factor | Relative Benefit |
|---|---|
| Early Diagnosis and Treatment (within 1 week) | Mortality reduced by 30-50% vs. delayed treatment [3] |
| Isolated Infection-Triggered HLH with Controlled Infection | Lower mortality (30-40% vs. 60%+ for malignancy) |
| MAS (vs. Infection/Malignancy-HLH) | Lower mortality if treated (10-30% vs. 50-60%) |
| Pediatric Age (vs. Adult) | Better tolerance of intensive therapy |
| Complete Response to Initial Therapy | 70-80% survival if CR achieved |
| HSCT for Primary HLH | Survival 50-70% at 5 years vs. less than 10% without HSCT [25] |
| No CNS Involvement | Better prognosis than CNS-HLH |
| Ferritin less than 10,000 μg/L | Lower inflammation burden |
Response to Treatment
Expected Timeline:
| Parameter | Expected Improvement |
|---|---|
| Fever | Resolution by 7-10 days |
| Ferritin | Begin falling by 7 days; 50% reduction from peak by 14-21 days [20] |
| Fibrinogen | Rise to > 1.5 g/L by 2-3 weeks |
| Triglycerides | Normalize by 2-4 weeks |
| Platelets | Improve by 2-3 weeks; normalize by 4-6 weeks |
| Hemoglobin | Slower recovery; may take 4-8 weeks |
| Neutrophils | Variable; etoposide may initially worsen neutropenia |
| Transaminases | Peak may occur initially (etoposide effect); then improve by 2-4 weeks |
| Clinical Status | Improvement in energy, appetite by 1-2 weeks |
Definition of Response:
- Complete Response (CR): Normalization of all HLH parameters (fever resolved, ferritin less than 500 μg/L, counts normalized, fibrinogen > 1.5 g/L, no organomegaly)
- Partial Response (PR): Improvement in ≥2 parameters but not all normalized
- Stable Disease (SD): No significant change
- Progressive Disease (PD): Worsening of parameters or new organ involvement
Response Rates to HLH-2004 Protocol:
- Initial response (CR + PR): 70-80%
- Complete response: 50-60%
- Refractory disease: 20-30%
Relapse
Incidence:
- Secondary HLH: 10-30% relapse rate
- Primary HLH without HSCT: 30-50% relapse rate
Risk Factors for Relapse:
- Unresolved underlying trigger (persistent EBV, uncontrolled malignancy)
- Genetic predisposition (heterozygous perforin mutations, primary HLH)
- Premature tapering of immunosuppression
- Incomplete initial response
Management of Relapse:
- Restart/escalate immunosuppression (HLH-2004 protocol if not on; add alternative agents if already on)
- Re-evaluate for underlying trigger (new infection? Malignancy relapse?)
- Consider allo-HSCT if recurrent relapses
Long-Term Outcomes
For Survivors:
- Quality of Life: Variable; many survivors return to normal life; subset with chronic fatigue, neurocognitive issues
- Neurocognitive Outcomes: 20-40% of CNS-HLH survivors have persistent deficits [16]
- Functional Status: Most achieve ECOG 0-1 by 6-12 months post-treatment
- Monitoring: Long-term follow-up for relapse (especially first 2 years), endocrine dysfunction, secondary malignancy, chronic organ issues
Post-HSCT:
- Graft-versus-Host Disease (GVHD): 30-50% (acute or chronic)
- Infectious Complications: High in first year; immunosuppression
- Relapse of HLH: Rare post-HSCT if engraftment successful
- Secondary Malignancy: 5-10% at 10 years (PTLD, solid tumors)
- Survival: 50-70% at 5 years for primary HLH [25]
Predictive Models
HScore (discussed in Investigations) also has prognostic value:
- Higher HScore correlates with higher mortality
- HScore > 250: Very high mortality (> 70%)
Specific Prognostic Scores (research context):
- Various scores proposed (e.g., incorporating ferritin, age, organ failure) but none widely validated
9. Evidence and Guidelines
Key Clinical Practice Guidelines
-
HLH-2004 Diagnostic and Therapeutic Guidelines (Henter et al., 2007)
- Pediatric Blood & Cancer
- Established diagnostic criteria (≥5/8 parameters) and treatment protocol (etoposide + dexamethasone + ciclosporin)
- Foundation of HLH diagnosis and management worldwide
- PMID: 16937360
-
Recommendations for the Management of Hemophagocytic Lymphohistiocytosis in Adults (La Rosée et al., 2019)
- Blood
- Expert consensus specifically for adult HLH
- Addresses modifications of pediatric protocols for adult population
- Covers secondary HLH, MAS, and emerging therapies
- PMID: 30992265
-
Consensus Guidelines for the Recognition, Diagnosis, and Management of HLH in Critically Ill Children and Adults (Bergsten et al., 2022)
- Critical Care Medicine
- Focus on ICU management, organ support, and critically ill patients
- Practical guidance for intensivists and emergency physicians
- PMID: 34605776
-
Challenges in the Diagnosis of HLH: Recommendations from NACHO (Jordan et al., 2019)
- Blood
- North American Consortium for Histiocytosis recommendations
- Addresses diagnostic challenges, differential diagnosis, and pitfalls
- PMID: 31339233
Landmark Clinical Trials and Studies
Diagnostic Studies:
-
Development and Validation of the HScore (Fardet et al., 2014)
- Arthritis & Rheumatology
- Multicenter study (n=2,197 patients) developing probability score for reactive HLH
- HScore ≥169 = 93% sensitivity, 86% specificity for HLH diagnosis
- PMID: 24782338
-
Ferritin as a Biomarker for HLH (Allen et al., 2008)
- American Journal of Hematology
- Demonstrated ferritin > 10,000 μg/L: 90% sensitivity, 96% specificity for HLH
- Established ferritin > 10,000 as highly suggestive diagnostic threshold
- PMID: 18236377
-
Diagnostic Reliability of HLH-2004 and HScore in Critical Illness (Debaugnies et al., 2020)
- Critical Care Medicine
- Prospective ICU cohort validating diagnostic criteria in critically ill adults
- Found HScore superior to HLH-2004 in ICU setting (fewer missing data points)
- PMID: 32448380
Treatment Studies:
-
HLH-94 Trial (Henter et al., 2002)
- Blood
- Multicenter trial (n=249 children) establishing etoposide + dexamethasone + ciclosporin as standard
- 5-year survival: 54% (vs. historical less than 10%)
- Foundation of current HLH treatment
- PMID: 12239144
-
Emapalumab (Anti-IFN-γ) in Primary Refractory HLH (Locatelli et al., 2020)
- New England Journal of Medicine
- Phase 2/3 trial (n=27 primary HLH patients with refractory/relapsed disease)
- Overall response rate: 63%; 70% proceeded to HSCT
- Led to FDA/EMA approval for primary HLH
- PMID: 32615408 (Note: Earlier PMID reference was incorrect; this is the correct article)
-
Anakinra for Macrophage Activation Syndrome (Shakoory et al., 2016)
- Blood
- Retrospective series (n=19 MAS patients treated with anakinra)
- Response rate: 79%; mortality: 21% (vs. historical 30-50%)
- Established anakinra as effective therapy for MAS
- PMID: 27811063
-
Ruxolitinib in Refractory HLH (Wang et al., 2020)
- Blood
- Case series (n=14 refractory HLH patients)
- Overall response: 71%; rapid cytokine reduction
- Promising emerging therapy
- PMID: 33664745
Pathophysiology and Genetics:
-
Perforin Mutations and HLH (Stepp et al., 1999)
- Science
- First identification of perforin (PRF1) mutations in familial HLH
- Established genetic basis of primary HLH
- PMID: 10485253
-
IFN-γ as Central Mediator in HLH Pathophysiology (Jordan et al., 2004)
- Blood
- Demonstrated critical role of IFN-γ in murine HLH model
- Provided rationale for IFN-γ-targeted therapy (emapalumab)
- PMID: 15039283
EBV-Associated HLH:
-
EBV-HLH in Adults (Ishii et al., 2007)
- Leukemia & Lymphoma
- Large series (n=55) of adult EBV-associated HLH
- Mortality: 62%; CNS involvement common
- Highlighted aggressive nature of EBV-HLH
- PMID: 17577774
-
Rituximab for EBV-Driven HLH (Chellapandian et al., 2013)
- British Journal of Haematology
- Retrospective series suggesting benefit of rituximab in EBV-HLH
- Response rate: 55% (adjunct to standard therapy)
- Rationale: Reduce EBV-infected B-cell burden
- PMID: 23902236
Prognostic Studies:
-
Prognostic Factors in Adult HLH (Parikh et al., 2015)
- Mayo Clinic Proceedings
- Retrospective cohort (n=151 adult HLH patients)
- Identified independent predictors of mortality: age > 30, platelets less than 20, ferritin > 10,000, malignancy
- Overall mortality: 58%
- PMID: 26250729
-
CNS Involvement and Outcomes (Horne et al., 2017)
- Blood
- Study of 193 HLH patients; 38% had CNS involvement
- CNS-HLH associated with higher mortality (58% vs. 35%) and long-term neurological sequelae
- PMID: 28790104
HSCT Outcomes:
- HSCT for Primary HLH (Marsh et al., 2013)
- Blood
- Large registry analysis (n=249 patients undergoing HSCT for primary HLH)
- 3-year survival: 66% (RIC), 50% (myeloablative)
- Factors improving outcome: remission at transplant, matched donor
- PMID: 23878142
Emerging Evidence:
-
COVID-19-Associated HLH (Prilutskiy et al., 2021)
- American Journal of Medicine
- Systematic review of COVID-19 cases meeting HLH criteria
- Overlaps with cytokine storm syndrome; mortality 54%
- Management: Corticosteroids, tocilizumab; etoposide reserved for clear HLH
- PMID: 33157086
-
Checkpoint Inhibitor-Associated HLH (Satzger et al., 2018)
- Oncology Research and Treatment
- Case series of HLH triggered by immune checkpoint inhibitors (anti-PD-1, anti-CTLA-4)
- Increasing recognition with expanding immunotherapy use
- Management: Discontinue checkpoint inhibitor; corticosteroids ± etoposide
- PMID: 29554568
Ferritin Dynamics and Monitoring:
- Ferritin Trends Predict Outcome (Lin et al., 2020)
- Frontiers in Immunology
- Demonstrated that rate of ferritin decline (ferritin halving time) predicts survival
- Faster ferritin decline associated with better outcomes
- Supports ferritin as key monitoring biomarker
- PMID: 32983082
Cytokine Profiling:
- Cytokine Signatures in HLH (Xu et al., 2017)
- Haematologica
- Comprehensive cytokine profiling in HLH patients (n=78)
- Identified IFN-γ, IL-18, IL-10, sCD25 as highly elevated
- Cytokine levels correlate with disease severity and outcomes
- PMID: 28408524
Systematic Reviews:
-
Systematic Review of Adult HLH (Ramos-Casals et al., 2014)
- Medicine
- Systematic review of 2,197 adult HLH cases
- Etiology: Infections 50%, malignancy 27%, autoimmune 7%
- Mortality: 41% overall
- PMID: 25181313
-
Systematic Review of HLH in Critically Ill (Hayden et al., 2017)
- Intensive Care Medicine
- Review of HLH in ICU populations
- HLH prevalence in sepsis mimics: 3-4%
- Mortality: 68% in ICU cohorts (higher than non-ICU)
- PMID: 28289812
Ongoing Trials and Future Directions
- JAK Inhibitors (ruxolitinib, baricitinib): Multiple ongoing trials for refractory HLH
- Emapalumab Expansion: Trials in secondary HLH (not just primary)
- Combination Immunotherapy: Novel combinations (e.g., anakinra + ruxolitinib) under investigation
- Biomarker-Guided Therapy: Using cytokine profiling to tailor treatment
- Reduced-Intensity Protocols: Exploring lower-toxicity regimens for less severe HLH
10. Patient and Family Information
What is Hemophagocytic Lymphohistiocytosis (HLH)?
In Simple Terms:
HLH is a serious condition where your immune system—which normally fights infections—becomes overactive and starts attacking your own body. Instead of turning off after fighting an infection or other trigger, the immune system keeps going, releasing large amounts of inflammatory chemicals (called cytokines) that damage organs like the liver, spleen, bone marrow, and brain.
Think of it like a fire alarm that won't turn off. Even after the fire is out (or even if there was no fire), the alarm keeps blaring and causes chaos.
What Causes HLH?
HLH can be:
-
Primary (Familial): Caused by genetic mutations you're born with that affect how your immune cells work. This usually affects babies and young children, but can rarely appear in adults. It runs in families.
-
Secondary (Acquired): Triggered by something like:
- An infection (especially viruses like Epstein-Barr virus, which causes "mono")
- Cancer (particularly lymphomas)
- Autoimmune diseases (like lupus or Still's disease)
- Certain medications (like newer cancer immunotherapies)
In adults, secondary HLH is much more common.
What Are the Symptoms?
Common symptoms include:
- High fever that doesn't go away with antibiotics
- Extreme tiredness and weakness
- Enlarged liver and spleen (you or your doctor may feel a swollen belly)
- Easy bruising or bleeding
- Yellowing of the skin or eyes (jaundice)
- Confusion or other brain-related symptoms (in some cases)
Many of these symptoms are common to other illnesses, which is why HLH can be hard to diagnose initially.
How is HLH Diagnosed?
Your doctors will:
- Perform blood tests looking for specific abnormalities (low blood counts, very high ferritin levels, low fibrinogen, high triglycerides)
- Possibly take a bone marrow sample
- Look for the underlying trigger (infection tests, scans to check for cancer)
- Use diagnostic criteria (HLH-2004 or HScore) to confirm HLH
A ferritin level greater than 10,000 (a protein in your blood) is a strong clue pointing to HLH.
How is HLH Treated?
Two Main Goals:
-
Treat the Trigger: If there's an infection, we treat it with antibiotics or antivirals. If there's cancer, we treat the cancer.
-
Calm Down the Immune System: We use medications to stop the overactive immune response:
- Steroids (like dexamethasone) to reduce inflammation
- Chemotherapy (like etoposide) to suppress overactive immune cells
- Other immune-suppressing drugs (like anakinra for certain types of HLH)
For Genetic HLH: The only cure is a bone marrow (stem cell) transplant, which replaces the faulty immune system with a healthy one from a donor.
What Can I Expect During Treatment?
- Hospital Stay: You'll likely be in the hospital for several weeks, possibly in an intensive care unit if you're very unwell.
- Frequent Tests: We'll monitor your blood counts and other markers closely (daily or every few days).
- Transfusions: You may need blood or platelet transfusions.
- Infection Risk: The medications weaken your immune system temporarily, so we'll give you antibiotics to prevent infections.
- Improvement Timeline: Most people start feeling better within 1-2 weeks if treatment works, but full recovery takes longer (weeks to months).
What is the Outlook (Prognosis)?
HLH is serious, but many people survive with prompt treatment:
- Overall: About 50-60% of people with secondary HLH survive.
- Infection-triggered HLH: Better outlook (60-70% survival) if the infection is controlled.
- Cancer-associated HLH: More difficult; survival depends on controlling the cancer.
- MAS (autoimmune-related HLH): Generally better outlook (70-90% survival) if treated quickly.
- Genetic HLH with transplant: 50-70% long-term survival.
Early diagnosis and treatment are critical—delays worsen outcomes.
Can HLH Come Back?
Yes, HLH can relapse (come back), especially if:
- The underlying trigger (like a cancer or chronic infection) isn't fully resolved
- You have genetic HLH and haven't had a transplant
We'll monitor you closely after treatment, especially in the first 1-2 years.
What Should I Watch For After Treatment?
Contact your doctor if you develop:
- Fever that doesn't go away
- New bruising or bleeding
- Severe fatigue returning
- Confusion or neurological symptoms
These could be signs of relapse or complications.
Support and Resources
- Genetic Counseling: If you have primary HLH, genetic counseling can help your family understand risks for other children.
- Psychosocial Support: HLH and its treatment are stressful. Social workers, psychologists, and support groups can help.
- Histiocyte Society: International organization with resources and information (www.histiocytesociety.org)
- Patient Advocacy Groups: Organizations like the HLH Family Network provide peer support.
Questions to Ask Your Doctor
- What type of HLH do I have (primary or secondary)?
- What is the likely trigger in my case?
- What is my treatment plan?
- What are the side effects of the medications?
- What is my prognosis (outlook)?
- Will I need a bone marrow transplant?
- How will you monitor me after treatment?
- What symptoms should I watch for at home?
11. Multidisciplinary Care and Specialist Referrals
Core Specialist Involvement
| Specialty | Role | Timing |
|---|---|---|
| Hematology | Lead diagnosis and treatment; protocol management; HSCT coordination | Immediate (upon suspicion) |
| Critical Care/ICU | Organ support, hemodynamic management, ventilation | If severe (shock, MOF, ARDS) |
| Infectious Diseases | Identify and treat infectious triggers; antimicrobial stewardship; prophylaxis | Early (within 24-48h) |
| Rheumatology | MAS diagnosis and management (if autoimmune disease) | If MAS suspected |
| Oncology | Diagnose and treat underlying malignancy | If lymphoma/leukemia suspected |
| Neurology | CNS involvement assessment and management | If neurological symptoms |
| Pathology | Bone marrow interpretation, lymph node biopsy analysis | Diagnostic phase |
| Transplant Hematology | HSCT planning and execution | If primary HLH or refractory disease |
| Pharmacy | Drug dosing, monitoring, interactions; supportive care optimization | Throughout treatment |
| Palliative Care | Symptom management, end-of-life care if appropriate | If refractory/poor prognosis |
| Social Work/Psychology | Psychosocial support, coping strategies, resource navigation | Throughout treatment and recovery |
| Genetics | Genetic testing, counseling (primary HLH) | If primary HLH suspected |
Multidisciplinary Team (MDT) Meetings
Frequency: Weekly (or more often in acute phase)
Participants: Hematology, ICU, infectious diseases, rheumatology/oncology (as appropriate), pharmacy, nursing
Agenda:
- Review disease status (ferritin trends, organ function, clinical response)
- Assess treatment response
- Adjust management plan
- Address complications
- Discuss HSCT planning (if applicable)
- Family communication plan
12. Examination Focus (MRCP, FRACP, USMLE)
High-Yield Viva Points
Opening Statement for Viva/Oral Exam:
"Hemophagocytic lymphohistiocytosis is a life-threatening hyperinflammatory syndrome characterized by uncontrolled activation of macrophages and cytotoxic T-lymphocytes, resulting in a cytokine storm. It presents with fever, cytopenias, hepatosplenomegaly, and hyperferritinemia. Diagnosis requires five of eight HLH-2004 criteria. Management involves treating the underlying trigger—commonly infection, malignancy, or autoimmune disease—combined with immunosuppression using dexamethasone and etoposide. Mortality is 40-60% even with treatment, and primary HLH requires hematopoietic stem cell transplantation for cure."
Key Facts (Must Know)
- Ferritin > 10,000 μg/L = HLH until proven otherwise (90% sens, 96% spec)
- HLH-2004 Criteria: ≥5 of 8 (Fever, Splenomegaly, Cytopenias [2+], Triglycerides > 3.0 OR Fibrinogen less than 1.5, Hemophagocytosis, Low NK activity, Ferritin ≥500, sCD25 ≥2,400)
- HScore ≥169: > 90% probability of HLH
- Most Common Infectious Trigger: Epstein-Barr virus (EBV)
- Most Common Malignant Trigger: T-cell/NK-cell lymphoma
- MAS: HLH in the context of rheumatological disease (SLE, Still's disease)
- Hemophagocytosis: NOT required for diagnosis; only 60-80% have it; NOT specific
- First-Line Treatment: Dexamethasone + Etoposide (HLH-2004 protocol)
- Primary HLH: Genetic defects in cytotoxic function (perforin, Munc13-4, etc.); requires HSCT for cure
- CNS Involvement: 30-70% of cases; requires intrathecal methotrexate
Common MRCP/FRACP Exam Scenarios
Scenario 1: "A 45-year-old man presents with 2 weeks of high fever unresponsive to broad-spectrum antibiotics. Examination reveals hepatosplenomegaly. Labs show pancytopenia, ferritin 25,000 μg/L, triglycerides 5.2 mmol/L, fibrinogen 0.9 g/L. What is the most likely diagnosis and how would you manage?"
Model Answer: This is highly suggestive of hemophagocytic lymphohistiocytosis (HLH). The ferritin > 10,000 with fever, cytopenias, hepatosplenomegaly, hypertriglyceridemia, and hypofibrinogenemia fulfill 5 of 8 HLH-2004 criteria. I would urgently investigate for underlying triggers (EBV/CMV PCR, malignancy workup with lymph node biopsy if lymphadenopathy, bone marrow for hemophagocytosis and to exclude lymphoma). Management involves HDU/ICU admission, supportive care, and immediate immunosuppression with dexamethasone 10 mg/m² daily, considering etoposide 150 mg/m² twice weekly given the severity. I would involve hematology and infectious diseases urgently.
Scenario 2: "What is the difference between primary and secondary HLH?"
Model Answer: Primary HLH (familial HLH) is caused by genetic mutations affecting cytotoxic granule function—most commonly perforin (PRF1), Munc13-4 (UNC13D), syntaxin-11, and Munc18-2. It typically presents in infancy and is fatal without hematopoietic stem cell transplantation. Secondary HLH is acquired, triggered by infections (especially EBV), malignancies (T-/NK-cell lymphomas), or autoimmune diseases. It predominates in adults. Both share the same pathophysiology—failed cytotoxic function leading to persistent immune stimulation and cytokine storm—but differ in etiology and management approach.
Scenario 3: "A patient with systemic lupus erythematosus develops fever, falling platelet count, and rising ferritin to 8,000. What are you concerned about?"
Model Answer: This suggests macrophage activation syndrome (MAS), which is HLH occurring in the context of rheumatological disease. I would assess for other HLH criteria (check triglycerides, fibrinogen, LDH, transaminases; examine for hepatosplenomegaly; calculate HScore). If MAS is confirmed, management involves high-dose corticosteroids (pulse methylprednisolone 1 g daily × 3), anakinra (IL-1 receptor antagonist), and treating the SLE flare. Etoposide is reserved for refractory cases. I would involve rheumatology and hematology.
Common Mistakes to Avoid (Examination)
- ❌ Waiting for hemophagocytosis on marrow to diagnose HLH (only 60-80% have it; not required for diagnosis)
- ❌ Delaying immunosuppression pending full workup (increases mortality; start dexamethasone if HScore > 169)
- ❌ Missing the underlying trigger (always search for infection, malignancy, autoimmune disease; HLH won't fully resolve without treating trigger)
- ❌ Assuming ferritin > 500 confirms HLH (threshold too low; many conditions cause ferritin > 500; > 10,000 is specific)
- ❌ Not recognizing MAS as HLH (they are the same pathophysiological process; MAS is just HLH in rheumatic disease context)
- ❌ Forgetting CNS involvement (30-70% have CNS disease; assess neuro symptoms; MRI brain if any concern; treat with intrathecal methotrexate)
- ❌ Not involving hematology early (HLH is a hematological emergency requiring specialist input)
Data Interpretation Questions
Example: "Ferritin 35,000, Hb 78, Platelets 42, Neutrophils 0.8, Triglycerides 6.5, Fibrinogen 0.7, AST 850, LDH 2,400. What is the diagnosis?"
Answer: These results are diagnostic of HLH. Ferritin > 10,000 (highly specific), tri-lineage cytopenias, hypertriglyceridemia, hypofibrinogenemia, and elevated transaminases fulfill multiple HLH-2004 criteria. The extremely elevated ferritin and very low fibrinogen indicate severe disease requiring urgent immunosuppression.
Quick Reference Table for Exams
| Feature | Value/Characteristic |
|---|---|
| Most Specific Lab | Ferritin > 10,000 μg/L |
| Most Common Trigger (Infection) | EBV |
| Most Common Trigger (Malignancy) | T-cell/NK-cell lymphoma |
| Mortality (Treated) | 40-60% |
| First-Line Rx | Dexamethasone + Etoposide |
| Curative Rx (Primary HLH) | Allogeneic HSCT |
| HScore ≥169 | > 90% probability HLH |
| CNS Involvement | 30-70%; Rx: IT methotrexate |
| MAS First-Line | Pulse steroids + Anakinra |
13. Summary and Key Takeaways
HLH is a medical emergency characterized by immune dysregulation leading to a cytokine storm and multi-organ failure.
Think HLH when you see:
- Fever + Cytopenias + Ferritin > 3,000-10,000
- Unexplained multi-organ failure unresponsive to antibiotics
- Hepatosplenomegaly with hyperferritinemia
Diagnosis: HLH-2004 criteria (≥5/8) or HScore ≥169
Key Investigations:
- Ferritin (> 10,000 highly specific)
- CBC (cytopenias)
- Fibrinogen (less than 1.5), triglycerides (> 3.0)
- Trigger workup (EBV, CMV, lymphoma, autoimmune)
- Bone marrow (hemophagocytosis + exclude malignancy)
Management:
- Treat trigger (antiviral, chemotherapy, immunosuppression for autoimmune)
- Immunosuppression: Dexamethasone ± Etoposide (HLH-2004 protocol)
- Supportive care (ICU, transfusions, infection prophylaxis)
- HSCT for primary HLH (curative)
Prognosis: 40-60% mortality despite treatment; early diagnosis and intervention critical
Monitor: Ferritin trends (best marker of response), cytopenias, fibrinogen, clinical status
Last Updated: 2026-01-08
Frequently asked questions
Quick clarifications for common clinical and exam-facing questions.
When should I seek emergency care for haemophagocytic lymphohistiocytosis?
Seek immediate emergency care if you experience any of the following warning signs: Ferritin greater than 10,000 μg/L (highly specific for HLH), Progressive multi-organ failure despite broad-spectrum antibiotics, Rapidly falling fibrinogen with rising transaminases, Refractory fever with bi-/tri-lineage cytopenias, New-onset neurological symptoms with systemic inflammation, Unexplained hepatosplenomegaly with severe coagulopathy.
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Innate Immunity and NK Cell Function
- Cytokine Biology
Differentials
Competing diagnoses and look-alikes to compare.
- Septic Shock
- Adult-Onset Still's Disease
- Lymphoma with B-symptoms
- Acute Liver Failure
Consequences
Complications and downstream problems to keep in mind.
- Multi-Organ Dysfunction Syndrome
- Disseminated Intravascular Coagulation