Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and irreversible fibrosing interstitial pneumonia of unkn... MRCP exam preparation.
What matters first
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and irreversible fibrosing interstitial pneumonia of unkn... MRCP exam preparation.
Acute exacerbation
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Acute exacerbation
- Rapidly declining FVC
- Severe hypoxia
- Pulmonary hypertension
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- Nonspecific Interstitial Pneumonia
- Chronic Hypersensitivity Pneumonitis
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Idiopathic Pulmonary Fibrosis
1. Overview
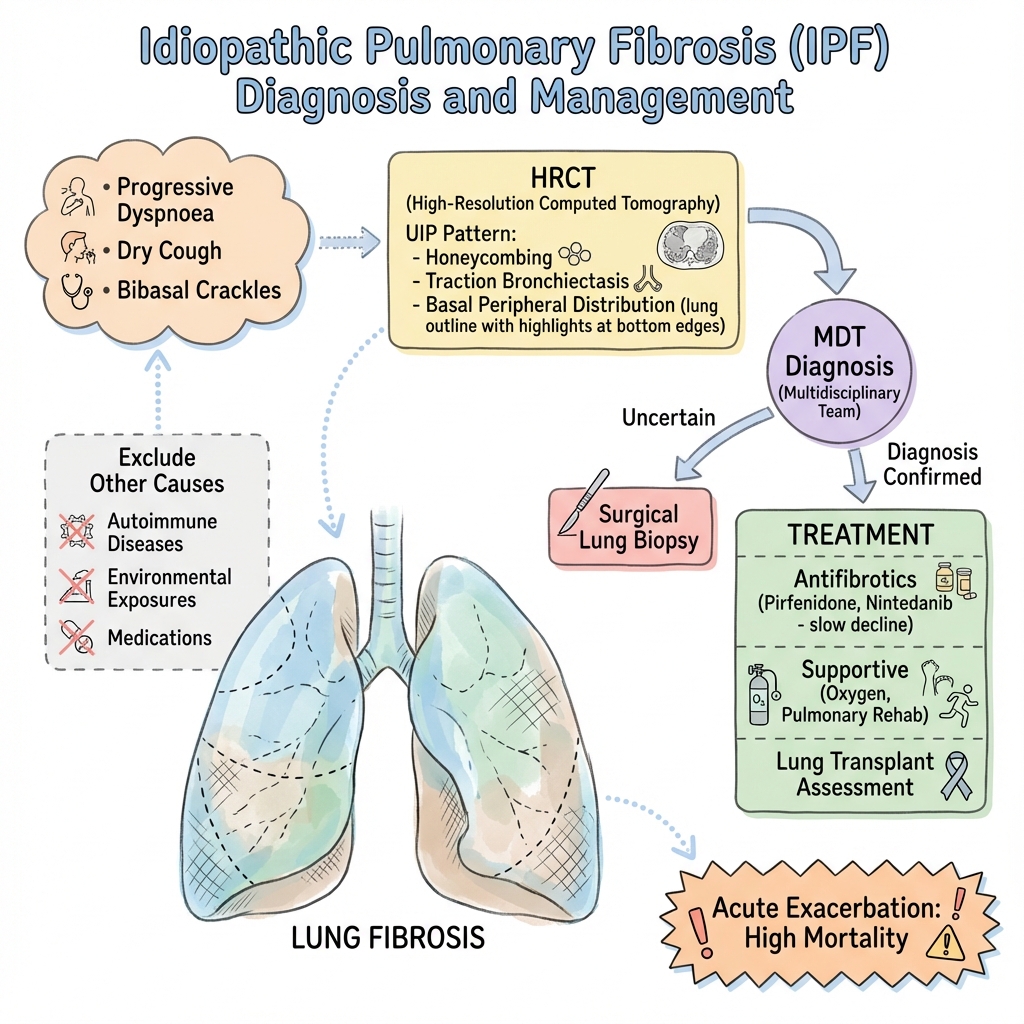
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and irreversible fibrosing interstitial pneumonia of unknown aetiology, occurring primarily in older adults and limited to the lungs. [1] It represents the most common and most lethal form of idiopathic interstitial pneumonia, characterised histologically by a pattern of usual interstitial pneumonia (UIP). [1,2] The disease is defined by the presence of UIP pattern on high-resolution computed tomography (HRCT) or surgical lung biopsy in the absence of alternative causes of interstitial lung disease. [1]
IPF is a disease of relentless fibrosis and progressive respiratory failure with a median survival of only 3-5 years from diagnosis, worse than many common cancers. [3] Patients typically present with insidious onset of progressive exertional dyspnoea and dry cough, often misdiagnosed initially as asthma or chronic obstructive pulmonary disease (COPD). The hallmark physical finding is bibasilar inspiratory crackles with a characteristic "Velcro" quality. [1] Clubbing is present in approximately 25-50% of patients and becomes more common with disease progression. [4]
The introduction of antifibrotic therapies—pirfenidone and nintedanib—has transformed IPF management, slowing disease progression by approximately 50% as measured by the rate of decline in forced vital capacity (FVC). [5,6] However, these agents do not reverse existing fibrosis or significantly improve survival, and lung transplantation remains the only potentially curative treatment for eligible patients. [7] Early recognition, accurate diagnosis through multidisciplinary discussion, and prompt initiation of antifibrotic therapy are critical to optimising outcomes.
2. Epidemiology
Incidence and Prevalence
The incidence of IPF varies widely by geography and methodology but is estimated at 3-9 cases per 100,000 person-years in Europe and North America, with higher rates reported in recent years likely due to improved recognition and an ageing population. [8] Prevalence estimates range from 14-43 per 100,000 population. [8,9] The disease is more common in men than women, with a male-to-female ratio of approximately 1.5-2:1. [9]
| Statistic | Value | Source |
|---|---|---|
| Annual incidence (Europe/North America) | 3-9 per 100,000 | [8] |
| Prevalence | 14-43 per 100,000 | [8,9] |
| Male:Female ratio | 1.5-2:1 | [9] |
| Median age at diagnosis | 66-70 years | [9,10] |
| Median survival from diagnosis | 3-5 years | [3] |
Demographics
IPF predominantly affects individuals over the age of 50 years, with the median age at diagnosis being 66-70 years. [9,10] The disease is rare before the age of 50, and diagnosis in younger individuals should prompt careful exclusion of alternative diagnoses such as familial interstitial pneumonia or connective tissue disease. [1]
Risk Factors
Environmental and Occupational Exposures:
- Cigarette smoking is the most consistently identified risk factor, with 60-75% of IPF patients having a history of smoking. [11] Current and former smokers have a significantly increased risk compared to never-smokers.
- Occupational exposures to metal dust, wood dust, agricultural dust, and livestock have been associated with increased IPF risk. [11]
- Viral infections (particularly herpesviruses such as Epstein-Barr virus and cytomegalovirus) have been implicated, though causality remains unproven. [12]
Genetic Factors:
- Familial pulmonary fibrosis accounts for approximately 5-20% of cases. [13]
- Common genetic variants (particularly in the MUC5B promoter region) increase IPF susceptibility and are present in approximately 35% of sporadic IPF cases versus 9% of controls. [14]
- Rare variants in genes encoding surfactant proteins and telomerase components have been identified in familial cases. [13]
Other Risk Factors:
- Gastroesophageal reflux disease (GERD) is highly prevalent (approximately 90%) in IPF patients and may contribute to disease pathogenesis through microaspiration. [15]
- Male sex and advanced age are consistent demographic risk factors.
3. Aetiology and Pathophysiology
Unknown Aetiology
Despite extensive research, the initiating trigger for IPF remains unknown. The current paradigm proposes that repetitive alveolar epithelial injury in genetically susceptible individuals leads to aberrant wound healing responses characterised by excessive fibroblast proliferation, myofibroblast differentiation, and pathological extracellular matrix deposition. [2,16]
Pathophysiological Mechanisms
Alveolar Epithelial Injury: The disease begins with injury to type II alveolar epithelial cells, the progenitor cells responsible for producing surfactant and regenerating the alveolar epithelium. [2] In IPF, these cells undergo apoptosis and fail to regenerate appropriately, leading to exposure of the basement membrane and activation of fibrotic pathways.
Aberrant Wound Healing: Unlike normal wound healing which terminates once tissue repair is complete, IPF is characterised by persistent and progressive fibrosis. [16] This aberrant response involves:
- Fibroblast Activation and Proliferation: Activated fibroblasts migrate to sites of epithelial injury and proliferate excessively.
- Myofibroblast Differentiation: Fibroblasts differentiate into myofibroblasts, which are contractile cells that produce excessive amounts of extracellular matrix proteins including collagen.
- Resistance to Apoptosis: IPF fibroblasts and myofibroblasts demonstrate resistance to apoptotic signals, allowing them to persist and continue producing matrix.
Molecular Mechanisms:
Transforming Growth Factor-β (TGF-β) Pathway: TGF-β is the central pro-fibrotic cytokine in IPF pathogenesis. [16] It is produced in an inactive form bound to latency-associated peptide (LAP) and must be activated by integrins (particularly αvβ6 integrin) expressed on damaged epithelial cells. Active TGF-β promotes:
- Fibroblast-to-myofibroblast differentiation
- Extracellular matrix protein synthesis
- Epithelial-to-mesenchymal transition (EMT)
- Inhibition of matrix metalloproteinases (MMPs) that would normally degrade excess matrix
Wnt/β-Catenin Signalling: Aberrant activation of Wnt/β-catenin signalling has been demonstrated in IPF lungs and contributes to fibroblast activation and proliferation. [17] This pathway normally regulates cell proliferation and differentiation during development but becomes reactivated in fibrotic disease.
Platelet-Derived Growth Factor (PDGF): PDGF is a potent mitogen for fibroblasts and contributes to their proliferation and migration to sites of injury. Nintedanib, one of the approved antifibrotic agents, targets PDGF receptors among others.
Fibroblast Growth Factor (FGF): FGF signalling promotes fibroblast proliferation and survival. Nintedanib also inhibits FGF receptors.
Coagulation Cascade: Activation of the coagulation cascade with thrombin generation has been implicated in IPF. Thrombin is a potent pro-fibrotic mediator that activates fibroblasts and promotes matrix deposition.
Cellular Senescence: Accumulation of senescent cells in IPF lungs contributes to disease pathogenesis through the senescence-associated secretory phenotype (SASP), which involves secretion of pro-inflammatory and pro-fibrotic mediators. [18]
Telomere Dysfunction: Short telomeres have been identified in familial and sporadic IPF cases and contribute to cellular senescence and impaired epithelial regeneration. [13]
Histopathological Pattern: Usual Interstitial Pneumonia (UIP)
The histopathological hallmark of IPF is the UIP pattern, characterised by:
- Heterogeneous Involvement: Alternating areas of normal lung, active fibrosis, and established fibrosis (honeycombing) with temporal heterogeneity—areas at different stages of fibrosis.
- Fibroblastic Foci: Collections of proliferating myofibroblasts at the advancing edge of fibrosis, representing sites of active disease.
- Honeycombing: Cystic airspaces lined by bronchiolar epithelium, representing end-stage fibrosis with architectural distortion.
- Subpleural and Paraseptal Distribution: Fibrosis predominantly affects the peripheral, subpleural regions and lower lobes.
- Minimal Inflammation: Unlike other forms of interstitial pneumonia, UIP demonstrates minimal lymphocytic inflammation.
4. Clinical Presentation
Symptoms
Cardinal Symptoms:
Progressive Exertional Dyspnoea: The most common presenting symptom, reported by over 90% of patients. [1] Dyspnoea is insidious in onset, gradually progressive over months to years, and characteristically worse with exertion. Patients often attribute symptoms to deconditioning or ageing, leading to delays in diagnosis. The rate of progression is variable but relentless.
Chronic Dry Cough: Present in 75-85% of patients. [4] The cough is typically non-productive and can be debilitating, significantly affecting quality of life. It is often refractory to conventional anti-tussive therapies.
Fatigue and Malaise: Non-specific but common complaints that contribute to reduced quality of life and functional capacity.
Associated Symptoms:
- Chest Discomfort: Vague chest tightness or discomfort may be reported but is not typically a prominent feature.
- Weight Loss: May occur in advanced disease.
- Symptoms of Complications: Ankle swelling suggesting right heart failure from pulmonary hypertension, or acute worsening suggesting acute exacerbation.
| Symptom | Frequency | Clinical Significance |
|---|---|---|
| Progressive dyspnoea | > 90% | Hallmark symptom; insidious onset |
| Dry cough | 75-85% | Often refractory to treatment |
| Fatigue | Common | Impacts quality of life |
| Inspiratory crackles | > 90% | Highly characteristic physical finding |
| Clubbing | 25-50% | More common with disease progression |
Signs
General Inspection:
- Patients may appear comfortable at rest in early disease but demonstrate tachypnoea and use of accessory muscles in advanced disease.
- Central cyanosis indicates severe hypoxaemia.
- Cachexia may be present in advanced disease.
Respiratory Examination:
Inspiratory Crackles: Fine, late inspiratory "Velcro-like" crackles are present in over 90% of IPF patients and represent the most characteristic physical finding. [1] Crackles are typically bilateral and bibasilar, reflecting the distribution of fibrosis. They are heard best at the posterior lung bases and may extend to the mid-zones as disease progresses. The "Velcro" quality refers to the distinctive sound resembling the separation of Velcro fasteners.
Clubbing: Digital clubbing is present in approximately 25-50% of patients at diagnosis and increases in frequency with disease duration. [4] Its pathogenesis in IPF is unclear but may relate to chronic hypoxia and circulating mediators.
Reduced Chest Expansion: Chest wall movement is reduced, reflecting restrictive lung physiology.
Cardiovascular Examination:
Signs of Pulmonary Hypertension and Cor Pulmonale:
- Elevated jugular venous pressure
- Right ventricular heave
- Loud P2 (pulmonary component of second heart sound)
- Tricuspid regurgitation murmur
- Peripheral oedema
- Hepatomegaly
These findings indicate advanced disease with development of secondary pulmonary hypertension.
Oxygen Desaturation: Pulse oximetry typically shows normal or near-normal oxygen saturation at rest in early disease but significant desaturation with exertion (6-minute walk test). Resting hypoxaemia indicates more advanced disease.
5. Differential Diagnosis
Accurate diagnosis of IPF requires exclusion of other causes of interstitial lung disease that may have similar clinical presentations but differ in treatment and prognosis. The differential diagnosis is broad and includes:
Idiopathic Interstitial Pneumonias (Other Than IPF)
1. Nonspecific Interstitial Pneumonia (NSIP):
- Key Distinguishing Features: NSIP presents with more uniform, temporally homogeneous fibrosis on HRCT and histology, with less honeycombing and more ground-glass opacification. [1] Prognosis is significantly better than IPF.
- HRCT Pattern: Ground-glass opacities, fine reticulation, possible subpleural sparing (unlike IPF).
- Clinical Context: May be idiopathic or associated with connective tissue disease (CTD).
2. Cryptogenic Organising Pneumonia (COP):
- Key Distinguishing Features: Subacute presentation (weeks to months), patchy consolidation on HRCT, excellent response to corticosteroids.
- HRCT Pattern: Consolidation with peribronchovascular and subpleural distribution, reverse halo sign.
3. Desquamative Interstitial Pneumonia (DIP):
- Key Distinguishing Features: Strong association with smoking, ground-glass opacities on HRCT, improvement with smoking cessation.
4. Acute Interstitial Pneumonia (AIP):
- Key Distinguishing Features: Acute presentation resembling acute respiratory distress syndrome (ARDS), diffuse alveolar damage on histology, high mortality.
Chronic Hypersensitivity Pneumonitis (CHP)
Key Distinguishing Features:
- History of antigen exposure (birds, mould, hot tubs) though often not identified (30-50% of cases).
- Upper and mid-lung predominance on HRCT (unlike IPF which is basal).
- Mosaic attenuation pattern, centrilobular nodules, and air trapping on expiratory imaging.
- Minimal honeycombing.
- The distinction from IPF can be challenging, particularly in fibrotic CHP. [19]
Connective Tissue Disease-Associated Interstitial Lung Disease (CTD-ILD)
Key Distinguishing Features:
- Features of underlying CTD: arthritis, skin changes (rashes, sclerodactyly), Raynaud's phenomenon, myositis, sicca symptoms.
- Positive autoantibodies: ANA, RF, anti-CCP, anti-Scl-70, anti-Ro, anti-La, anti-Jo-1, and others.
- May have UIP or NSIP pattern.
- Conditions include: systemic sclerosis, rheumatoid arthritis, polymyositis/dermatomyositis, Sjögren's syndrome, systemic lupus erythematosus.
- Important to screen for CTD in all patients with suspected IPF, particularly younger patients and women.
Sarcoidosis
Key Distinguishing Features:
- Multisystem disease (lymph nodes, skin, eyes, heart).
- Hilar and mediastinal lymphadenopathy on chest imaging.
- Upper and mid-lung predominance of parenchymal disease.
- Perilymphatic nodularity on HRCT.
- Noncaseating granulomas on biopsy.
- Younger age group (typically 20-40 years).
Occupational and Environmental Lung Diseases
Asbestosis:
- History of asbestos exposure (often occupational: shipyards, construction, insulation).
- Pleural plaques on imaging.
- Basal predominant fibrosis similar to IPF.
- Exclusion based on exposure history.
Silicosis:
- Occupational exposure to silica (mining, sandblasting, quarrying).
- Upper lobe predominance, nodular pattern.
- Eggshell calcification of lymph nodes.
Coal Worker's Pneumoconiosis:
- Coal mining exposure.
- Upper lobe nodularity, progressive massive fibrosis.
Drug-Induced Interstitial Lung Disease
Key Drugs Associated with Pulmonary Fibrosis:
- Chemotherapy agents: bleomycin, methotrexate, nitrosoureas.
- Antiarrhythmics: amiodarone.
- Antibiotics: nitrofurantoin.
- Immunosuppressants: azathioprine.
Key Distinguishing Features:
- Temporal relationship to drug exposure.
- Improvement upon drug withdrawal.
- Variable HRCT patterns.
Radiation-Induced Lung Injury
- History of thoracic radiation therapy.
- Geographic distribution corresponding to radiation field.
- Temporal relationship (acute: 1-3 months; chronic: > 6 months post-radiation).
| Differential Diagnosis | Key Distinguishing Features |
|---|---|
| NSIP | Ground-glass opacities, subpleural sparing, better prognosis |
| Chronic Hypersensitivity Pneumonitis | Upper/mid-lung predominance, mosaic attenuation, antigen exposure history |
| CTD-ILD | Extrapulmonary CTD features, positive autoantibodies |
| Sarcoidosis | Lymphadenopathy, upper lobe predominance, younger age, granulomas |
| Asbestosis | Asbestos exposure history, pleural plaques |
| Drug-Induced ILD | Temporal relationship to drug exposure |
6. Investigations
The diagnosis of IPF requires integration of clinical, radiological, and pathological findings through multidisciplinary discussion (MDD) involving pulmonologists, radiologists, and pathologists. [1]
First-Line Investigations
High-Resolution Computed Tomography (HRCT) of the Chest:
HRCT is the key investigation for diagnosing IPF and can obviate the need for surgical lung biopsy in patients with typical findings. [1] The ATS/ERS/JRS/ALAT 2018 guidelines define four HRCT patterns of UIP with different diagnostic certainties:
UIP Pattern (Definite UIP):
If all four features are present, this indicates a UIP pattern:
- Subpleural and basal predominance of fibrosis
- Honeycombing with or without peripheral traction bronchiectasis or bronchiolectasis
- Reticular pattern
- Absence of features inconsistent with UIP (see below)
Probable UIP Pattern:
Present when there is:
- Subpleural and basal predominant reticular pattern
- Traction bronchiectasis or bronchiolectasis
- Absence of honeycombing
- Absence of features inconsistent with UIP
Indeterminate for UIP:
Patterns that do not clearly fit UIP or alternative diagnoses:
- Subtle reticulation
- Cannot exclude features of alternative diagnosis
Features Inconsistent with UIP (Suggest Alternative Diagnosis):
- Predominant ground-glass opacification
- Predominant consolidation
- Mosaic attenuation/air trapping (suggests CHP)
- Centrilobular nodules (suggests CHP, respiratory bronchiolitis)
- Diffuse cysts (suggests lymphangioleiomyomatosis, Langerhans cell histiocytosis)
- Upper or mid-lung predominance
- Peribronchovascular predominance
Critical Point: In the appropriate clinical context (older adult, no exposure history, no CTD features), a definite UIP pattern on HRCT is sufficient for IPF diagnosis without requiring surgical lung biopsy. [1]
Pulmonary Function Tests (PFTs):
Spirometry and Lung Volumes:
- Restrictive pattern: Reduced FVC, reduced total lung capacity (TLC), preserved or elevated FEV1/FVC ratio.
- FVC is used as a key marker of disease severity and progression.
Diffusing Capacity (DLCO):
- Reduced DLCO, often disproportionately reduced compared to lung volumes.
- DLCO impairment reflects both loss of alveolar-capillary units and V/Q mismatch.
Gas Exchange:
- Arterial blood gas may show reduced PaO2, widened A-a gradient, particularly with exercise.
- Hypoxaemia at rest indicates advanced disease.
Exercise Testing (6-Minute Walk Test):
- Reduced exercise tolerance.
- Oxygen desaturation during exercise.
- Reduced 6-minute walk distance.
- The extent of desaturation and distance walked have prognostic implications.
| PFT Parameter | Typical Findings in IPF |
|---|---|
| FVC | Reduced (restrictive pattern) |
| TLC | Reduced |
| FEV1/FVC | Normal or elevated (> 0.7) |
| DLCO | Reduced (often severely) |
| PaO2 | Normal early; reduced in advanced disease |
Blood Tests:
Autoimmune Screening: Essential to exclude CTD-ILD:
- Antinuclear antibody (ANA)
- Rheumatoid factor (RF)
- Anti-cyclic citrullinated peptide (anti-CCP)
- Myositis panel (anti-Jo-1, anti-Scl-70, anti-synthetase antibodies)
- Anti-Ro, anti-La (Sjögren's syndrome)
A comprehensive autoimmune screen should be performed in all patients, particularly those who are younger, female, or have suggestive clinical features.
Other Blood Tests:
- Full blood count (FBC)
- Renal and liver function
- Serum ACE and calcium (elevated in sarcoidosis, though not sensitive)
Serum Biomarkers:
- Several biomarkers (KL-6, SP-A, SP-D, MMP-7) have been studied but are not routinely used in clinical practice for diagnosis.
Second-Line Investigations
Surgical Lung Biopsy:
Indicated when:
- HRCT shows probable UIP, indeterminate, or alternative pattern AND
- The patient is a suitable surgical candidate AND
- The result would change management
Technique:
- Video-assisted thoracoscopic surgery (VATS) is preferred over open biopsy due to lower morbidity.
- Multiple biopsies from different lobes (typically lower lobe and either lingula or middle lobe) are recommended to account for spatial heterogeneity.
Histopathological UIP Pattern:
- Required features: dense fibrosis with architectural distortion (honeycombing), patchy involvement, fibroblastic foci.
- Temporally heterogeneous fibrosis (areas of old and new fibrosis).
- Subpleural/paraseptal predominance.
Bronchoscopy with Bronchoalveolar Lavage (BAL) and Transbronchial Biopsy:
Bronchoscopy is NOT required for IPF diagnosis but may be useful to exclude alternative diagnoses:
- BAL cell differential: lymphocytosis suggests CHP, NSIP, or CTD-ILD; eosinophilia suggests eosinophilic pneumonia or drug reaction.
- Transbronchial biopsy is rarely diagnostic for IPF due to small sample size but may identify granulomas (sarcoidosis), organising pneumonia, or infection.
Echocardiography:
Not required for diagnosis but recommended for:
- Assessment of cardiac function
- Screening for pulmonary hypertension (elevated right ventricular systolic pressure, right heart changes)
- Evaluation before lung transplant assessment
Right Heart Catheterisation:
Gold standard for diagnosing pulmonary hypertension if suspected on echocardiography or if being considered for lung transplantation.
Multidisciplinary Discussion (MDD)
The cornerstone of IPF diagnosis is MDD involving respiratory physicians, thoracic radiologists, and histopathologists (if biopsy performed). [1] MDD has been shown to improve diagnostic accuracy and confidence compared to individual assessments. The integrated clinical-radiological-pathological diagnosis is more accurate than any single modality alone.
Diagnostic Algorithm Based on ATS/ERS/JRS/ALAT 2018 Guidelines:
Step 1: Exclude Known Causes of ILD
- Obtain detailed history of environmental/occupational exposures, medications, CTD symptoms
- Perform autoimmune screening
Step 2: Obtain HRCT
- Classify pattern as: UIP, Probable UIP, Indeterminate, or Alternative diagnosis
Step 3: Consider Need for Surgical Lung Biopsy
| HRCT Pattern | Clinical Context | Diagnosis | Biopsy Needed? |
|---|---|---|---|
| UIP pattern | Typical (age > 60, no exposures, no CTD) | IPF | No |
| UIP pattern | Atypical features | Uncertain | Consider |
| Probable UIP | Typical | IPF (with confidence) | No (in many cases) |
| Probable UIP | Atypical | Uncertain | Yes |
| Indeterminate | Any | Uncertain | Yes (if suitable candidate) |
| Alternative pattern | Any | Not IPF | No (for IPF diagnosis) |
Step 4: Multidisciplinary Discussion
- Integrate clinical, radiological, and pathological (if available) data
- Reach consensus diagnosis
- Assign level of diagnostic confidence
7. Classification and Prognostication
Diagnostic Classification
IPF is classified as an idiopathic interstitial pneumonia (IIP), specifically the most common and severe form characterised by the UIP pattern. The 2013 ATS/ERS classification of IIPs distinguishes IPF from other IIPs based on histopathological and radiological patterns. [1]
Severity and Prognostic Staging
Several staging systems and prognostic indices have been developed to predict outcomes in IPF:
GAP Index (Gender-Age-Physiology):
The GAP index is a validated clinical prediction model that uses four variables to predict mortality risk: [20]
| Variable | Points |
|---|---|
| Gender | |
| Female | 0 |
| Male | 1 |
| Age (years) | |
| ≤60 | 0 |
| 61-65 | 1 |
| > 65 | 2 |
| FVC (% predicted) | |
| > 75% | 0 |
| 50-75% | 1 |
| less than 50% | 2 |
| DLCO (% predicted) | |
| > 55% | 0 |
| 36-55% | 1 |
| ≤35% or cannot perform | 2 |
Total Score: 0-8 points
GAP Stages:
- Stage I (0-3 points): 1-year mortality 6%, 3-year mortality 16%
- Stage II (4-5 points): 1-year mortality 16%, 3-year mortality 33%
- Stage III (6-8 points): 1-year mortality 39%, 3-year mortality 62%
Prognostic Factors:
Poor Prognostic Indicators:
- Male sex
- Advanced age
- Severe physiological impairment (low FVC, low DLCO)
- Extensive fibrosis on HRCT
- Definite UIP pattern (worse than probable UIP)
- Reduced 6-minute walk distance
- Severe oxygen desaturation during exercise
- Pulmonary hypertension
- Acute exacerbation
- Elevated serum biomarkers (e.g., KL-6, SP-D, though not routinely used)
Disease Behaviour:
IPF demonstrates variable disease behaviour with three broad phenotypes:
- Slow progressors: Gradual decline over years
- Rapid progressors: Steep decline in lung function over months
- Acute exacerbators: Sudden accelerated deterioration (discussed in Complications)
Unfortunately, predicting which patients will follow which trajectory remains challenging at diagnosis.
8. Management
The goals of IPF management are to slow disease progression, alleviate symptoms, maintain quality of life, prevent acute exacerbations, and identify suitable candidates for lung transplantation. [1]
Multidisciplinary Care
IPF patients should be managed in specialist interstitial lung disease centres with access to multidisciplinary teams including respiratory physicians, radiologists, pathologists, specialist nurses, physiotherapists, palliative care, and transplant services.
Antifibrotic Therapy
Two antifibrotic agents are approved for IPF and recommended by international guidelines for patients with mild-to-moderate disease (FVC ≥50% predicted): [1,5,6]
1. Pirfenidone:
Mechanism of Action: Pirfenidone has multiple anti-fibrotic, anti-inflammatory, and antioxidant effects including inhibition of TGF-β synthesis and activity, reduction of fibroblast proliferation, and reduction of extracellular matrix production.
Dosing:
- Target dose: 801 mg three times daily (2403 mg/day total)
- Titration schedule: Start 267 mg TDS for 7 days, then 534 mg TDS for 7 days, then 801 mg TDS maintenance
- Taken with food to reduce gastrointestinal side effects
Evidence: The ASCEND trial (2014) demonstrated that pirfenidone reduced the decline in FVC by approximately 50% compared to placebo (−164.9 mL vs −427.9 mL) over 52 weeks. [5] Pooled analysis of ASCEND and prior CAPACITY trials showed reduced all-cause mortality and improved progression-free survival.
Adverse Effects:
- Gastrointestinal: nausea, dyspepsia, anorexia, weight loss (most common, affecting 30-40%)
- Photosensitivity rash (20-30%): advise sun protection, avoid prolonged sun exposure
- Fatigue, dizziness
- Elevated liver enzymes (monitor LFTs monthly for 6 months then 3-monthly)
Monitoring:
- LFTs at baseline, monthly for 6 months, then every 3 months
- FBC, renal function at baseline and periodically
- Weight monitoring
- Counsel on sun protection
2. Nintedanib:
Mechanism of Action: Nintedanib is an intracellular tyrosine kinase inhibitor targeting multiple pro-fibrotic pathways including:
- Platelet-derived growth factor receptors (PDGFR α, β, γ)
- Fibroblast growth factor receptors (FGFR 1, 2, 3)
- Vascular endothelial growth factor receptors (VEGFR 1, 2, 3)
Dosing:
- Standard dose: 150 mg twice daily
- Reduced dose (100 mg twice daily) for tolerability if needed
- Taken with food
Evidence: The INPULSIS-1 and INPULSIS-2 trials (2014) demonstrated that nintedanib reduced the annual rate of FVC decline by approximately 50% compared to placebo (−114.7 mL/year vs −239.9 mL/year). [6] Subsequent studies confirmed effects across disease severity spectrum and in patients with progressive fibrosing ILDs beyond IPF.
Adverse Effects:
- Diarrhoea (60-70%): the most common side effect; often manageable with loperamide, dose reduction, or temporary interruption
- Nausea, vomiting, abdominal pain
- Elevated liver enzymes (monitor LFTs)
- Bleeding risk (VEGF inhibition affects clotting)
- Weight loss
Monitoring:
- LFTs at baseline, monthly for 3 months, then periodically
- Consider anticoagulation bleeding risk
- Advise loperamide for diarrhoea management
Choice Between Pirfenidone and Nintedanib:
Both agents have similar efficacy in slowing FVC decline. Choice is based on:
- Side effect profile: gastrointestinal upset common with both; diarrhoea more prominent with nintedanib, photosensitivity unique to pirfenidone
- Contraindications: nintedanib contraindicated in recent myocardial infarction, bleeding risk
- Patient preference and tolerability
- Cost and availability
Combination Therapy: Combination pirfenidone and nintedanib has been studied but is not recommended due to increased adverse events without clear additional efficacy benefit.
Duration of Treatment: Antifibrotic therapy should be continued indefinitely unless:
- Unacceptable side effects
- Disease progression to advanced stage where transplant is not an option and focus shifts to palliative care
- Lung transplantation (antifibrotics are typically discontinued post-transplant)
| Drug | Dose | Mechanism | Key Evidence | Common Side Effects |
|---|---|---|---|---|
| Pirfenidone | 801mg TDS | Anti-TGF-β, anti-proliferative | ASCEND: 50% reduction in FVC decline [5] | Nausea, photosensitivity, fatigue |
| Nintedanib | 150mg BD | Tyrosine kinase inhibitor (PDGFR, FGFR, VEGFR) | INPULSIS: 50% reduction in FVC decline [6] | Diarrhoea, nausea, liver enzyme elevation |
Therapies NOT Recommended
The following therapies have been studied and are NOT recommended for IPF based on lack of efficacy or harm: [1]
- Corticosteroids (alone or in combination with azathioprine): No benefit, increased risk of death and hospitalisation
- N-acetylcysteine (NAC): No benefit in the PANTHER-IPF trial
- Anticoagulation (warfarin): Increased mortality in the ACE-IPF trial
- Immunosuppressants (azathioprine, cyclophosphamide): No benefit, increased harm
- Endothelin receptor antagonists (bosentan, macitentan, ambrisentan): No benefit
Supportive and Symptomatic Management
Oxygen Therapy:
- Supplemental oxygen for patients with resting hypoxaemia (SpO2 less than 88-90%) or significant exertional desaturation
- Improves exercise tolerance and quality of life
- Ambulatory oxygen for patients who desaturate with exertion
- No evidence for survival benefit in IPF (unlike COPD)
Pulmonary Rehabilitation:
- Structured exercise training, education, and psychosocial support
- Improves exercise capacity, dyspnoea, and quality of life in IPF [21]
- Should be offered to all patients with symptomatic IPF
Cough Management:
- Chronic cough is debilitating and often refractory to treatment
- Trial therapies: inhaled corticosteroids (limited evidence), opiates (codeine, morphine), gabapentin, speech therapy techniques
- Management of GERD may help in some patients
Management of Gastroesophageal Reflux Disease (GERD):
- GERD is highly prevalent in IPF (up to 90%) and may contribute to disease progression through microaspiration
- Proton pump inhibitors (PPIs) or H2 antagonists
- Lifestyle modifications: head of bed elevation, avoid late meals, weight loss
- Evidence for anti-reflux therapy slowing IPF progression is limited
Vaccinations:
- Annual influenza vaccination
- Pneumococcal vaccination (PCV13 and PPSV23)
- COVID-19 vaccination
- Reduce risk of respiratory infections that can trigger acute exacerbations
Palliative Care:
- Early involvement of palliative care services for symptom management, advance care planning, and end-of-life discussions
- Opiates for refractory dyspnoea
- Anxiolytics for anxiety and panic
- Discussion of resuscitation status, intubation preferences, and place of death
Psychological Support:
- IPF has significant psychological burden: anxiety, depression, fear
- Access to psychological support, counselling, and support groups
Lung Transplantation
Lung transplantation is the only treatment that improves survival in IPF. [7] Median survival post-transplant is approximately 5-6 years, significantly better than untreated IPF.
Indications for Transplant Referral:
Patients should be referred early (at diagnosis or soon after) if they meet the following criteria:
- Confirmed IPF diagnosis
- Evidence of disease progression (declining FVC, worsening symptoms, declining functional status)
- Age typically less than 65-70 years (varies by transplant centre)
- Absence of absolute contraindications
Timing of Listing:
Patients should be listed for transplantation when they meet any of:
- FVC less than 80% predicted or DLCO less than 40% predicted
-
10% decline in FVC over 6 months
- Desaturation to less than 88% or > 50m decrease in 6-minute walk distance over 6 months
- Pulmonary hypertension
Absolute Contraindications:
- Active malignancy (within 2-5 years depending on type)
- Severe coronary artery disease not amenable to revascularisation
- Severe irreversible dysfunction of another major organ (kidney, liver, heart)
- Active or recent smoking (most centres require 6 months abstinence)
- Inability to comply with medical regimen
- Active substance abuse
- Severe obesity or severe malnutrition
- HIV infection (relative at some centres)
- Hepatitis B or C with histological liver damage
- Significant chest wall or spinal deformity
Relative Contraindications:
- Age > 65-70 years (varies by centre)
- Borderline organ dysfunction
- Colonisation with resistant or virulent organisms
- Severe osteoporosis
- Obesity (BMI > 30-35)
Type of Transplant:
- Bilateral sequential lung transplantation is standard for IPF
- Single lung transplantation is performed less commonly
Outcomes:
- 1-year survival: approximately 85-90%
- 5-year survival: approximately 50-60%
- 10-year survival: approximately 30%
- Complications: rejection, infection, bronchiolitis obliterans syndrome
Early referral is critical as waiting times can be long, and disease progression may render patients too ill for transplant if referred late.
9. Complications
Acute Exacerbation of IPF
Acute exacerbation is a sudden, clinically significant deterioration in IPF characterised by acute worsening of dyspnoea (typically within 30 days), new bilateral ground-glass opacities on HRCT superimposed on UIP pattern, and absence of identifiable cause (infection, heart failure, pulmonary embolism, pneumothorax, or other). [22]
Epidemiology:
- Occurs in 5-10% of IPF patients per year
- Cumulative incidence increases with time
- Precipitating factors often not identified but may include infection, aspiration, procedures (bronchoscopy, surgery)
Clinical Presentation:
- Acute onset or worsening dyspnoea (over days to weeks)
- Worsening hypoxaemia requiring increased oxygen or mechanical ventilation
- New bilateral radiographic infiltrates
Investigations:
- HRCT: new bilateral ground-glass opacities and/or consolidation superimposed on UIP pattern
- Exclusion of infection (sputum cultures, viral PCR, bronchoscopy if safe), heart failure (BNP, echocardiography), pulmonary embolism (CTPA)
- Worsening restrictive defect on PFTs if measurable
Management:
- Supportive care: supplemental oxygen, mechanical ventilation if required
- Corticosteroids: commonly used (e.g., methylprednisolone 500-1000 mg IV daily for 3 days followed by oral prednisolone taper) despite limited evidence of benefit
- Consideration of immunosuppression (cyclophosphamide) in some cases, though evidence is weak
- Antifibrotic therapy continuation if already on treatment
- Treatment of any identified triggers (antibiotics if infection suspected)
- Consideration of lung transplantation if eligible
Prognosis:
- In-hospital mortality: 50-60%
- Median survival after acute exacerbation: 3-4 months
- Acute exacerbation is a leading cause of death in IPF
Prevention:
- No proven strategies, but vaccination and avoidance of respiratory infections may help
- Caution with invasive procedures
Pulmonary Hypertension
Epidemiology:
- Develops in 30-50% of IPF patients, particularly those with advanced disease
- Defined as mean pulmonary artery pressure ≥20 mmHg on right heart catheterisation (updated from previous threshold of ≥25 mmHg)
Pathophysiology:
- Multifactorial: hypoxic pulmonary vasoconstriction, loss of vascular bed due to fibrosis, pulmonary vascular remodelling, left heart dysfunction
Clinical Presentation:
- Progressive dyspnoea (out of proportion to lung function decline)
- Signs of right heart failure: elevated JVP, peripheral oedema, hepatomegaly
- Reduced exercise tolerance
Investigations:
- Echocardiography: elevated right ventricular systolic pressure, right heart enlargement, tricuspid regurgitation
- Right heart catheterisation: gold standard for diagnosis
Management:
- Oxygen therapy for hypoxaemia
- Treatment of underlying IPF
- Pulmonary vasodilators have not been shown to benefit IPF-associated pulmonary hypertension and may cause harm; therefore, they are generally not recommended
- Lung transplantation
Prognosis:
- Presence of pulmonary hypertension is associated with significantly worse survival in IPF
Pneumothorax
- Spontaneous pneumothorax can occur due to rupture of subpleural cysts/honeycombing
- Higher risk with advanced disease
- Management challenging due to underlying lung disease
- May require chest tube drainage
- High recurrence rate
Lung Cancer
- IPF patients have increased risk of lung cancer (8-15% of patients)
- Most commonly squamous cell carcinoma and adenocarcinoma
- Risk factors: smoking, fibrosis itself (field effect)
- Predominantly occurs in areas of fibrosis (lower lobes peripherally)
- HRCT surveillance may detect early lesions
- Management challenging due to poor baseline lung function limiting surgical options
Respiratory Failure
- Progressive hypoxaemic respiratory failure is the usual cause of death in IPF
- Chronic type I respiratory failure (hypoxaemia without hypercapnia)
- Type II respiratory failure (hypoxaemia and hypercapnia) may develop in advanced disease or acute exacerbation
- Management: supplemental oxygen, mechanical ventilation (invasive or non-invasive) in selected cases
- Prognostic discussions regarding intubation and mechanical ventilation are essential
Venous Thromboembolism
- Increased risk of deep vein thrombosis (DVT) and pulmonary embolism (PE) in IPF patients, particularly during hospitalisations
- Prophylaxis with low-molecular-weight heparin during hospitalisations
| Complication | Frequency | Prevention | Management |
|---|---|---|---|
| Acute exacerbation | 5-10% per year | Vaccination, avoid respiratory infections | Corticosteroids, supportive care, mortality > 50% |
| Pulmonary hypertension | 30-50% | Oxygen therapy | Treat underlying IPF, transplant |
| Pneumothorax | Uncommon but increased risk | None specific | Chest tube drainage |
| Lung cancer | 8-15% | Smoking cessation | Limited by poor lung function |
| Respiratory failure | Universal eventually | Oxygen, pulmonary rehab | Supportive, palliative, transplant |
10. Prognosis
Natural History
IPF is a progressive and ultimately fatal disease. Without treatment, median survival from diagnosis is approximately 3-5 years, comparable to many aggressive malignancies. [3] However, the disease course is highly variable:
- Slow progressors (approximately 30%): Gradual decline in lung function over many years
- Intermediate progressors (approximately 50%): Steady decline over 3-5 years
- Rapid progressors (approximately 20%): Steep decline within 1-2 years
Predicting individual patient trajectory at diagnosis remains challenging.
Impact of Antifibrotic Therapy
Pirfenidone and nintedanib reduce the rate of FVC decline by approximately 50% but do not halt progression or reverse fibrosis. [5,6] Long-term observational data suggest possible survival benefit, though randomised trials were not powered to detect mortality differences.
Causes of Death
- Respiratory failure due to progressive fibrosis (most common)
- Acute exacerbation of IPF
- Cardiovascular disease (ischaemic heart disease, heart failure)
- Lung cancer
- Pulmonary embolism
- Infections (pneumonia)
Factors Affecting Prognosis
Baseline Severity:
- Worse lung function (low FVC, low DLCO) at diagnosis predicts poorer survival
- Extensive fibrosis on HRCT associated with worse outcomes
Disease Progression:
- Rapid decline in FVC (≥10% over 6 months) indicates poor prognosis
- Development of pulmonary hypertension worsens survival significantly
Acute Exacerbations:
- Acute exacerbations dramatically worsen prognosis with > 50% mortality and median survival of 3-4 months after event [22]
Comorbidities:
- Cardiovascular disease, diabetes, GERD, emphysema affect outcomes
GAP Index: As discussed in Classification section, GAP index stratifies patients into three mortality risk groups. [20]
Quality of Life
IPF significantly impairs health-related quality of life due to progressive dyspnoea, chronic cough, fatigue, psychological distress, and reduced functional capacity. Interventions such as pulmonary rehabilitation, symptom management, and psychological support are critical.
11. Prevention and Screening
Primary Prevention
No specific primary prevention strategies exist for IPF given its unknown aetiology. However, modifiable risk factors should be addressed:
- Smoking cessation: The most important modifiable risk factor
- Avoidance of occupational/environmental exposures: Appropriate respiratory protection in at-risk occupations
- GERD management: May reduce microaspiration risk, though impact on IPF development is unproven
Screening
There is no role for population-based screening for IPF given its relative rarity. However, surveillance in high-risk groups may be considered:
- Familial pulmonary fibrosis families: First-degree relatives may undergo periodic HRCT and PFT screening
- Genetic variants: Individuals with MUC5B polymorphism or telomerase mutations may be monitored, though clinical utility is uncertain
12. Key Guidelines
ATS/ERS/JRS/ALAT Clinical Practice Guideline (2018)
The definitive international guideline for IPF diagnosis and management. [1]
Key Recommendations:
- Diagnosis requires exclusion of other causes of ILD and presence of UIP pattern on HRCT or surgical lung biopsy
- Definite UIP pattern on HRCT in appropriate clinical context is sufficient for diagnosis without biopsy
- Multidisciplinary discussion is essential for accurate diagnosis
- Conditional recommendation FOR use of pirfenidone in mild-moderate IPF
- Conditional recommendation FOR use of nintedanib in mild-moderate IPF
- Strong recommendation AGAINST corticosteroid monotherapy, immunosuppression, anticoagulation
NICE Guideline (2013, updated 2017): Diagnosis and Management of Suspected Idiopathic Pulmonary Fibrosis
- Recommends referral of patients with suspected ILD to specialist ILD centres
- Supports use of pirfenidone and nintedanib for IPF patients with FVC 50-80% predicted
Examination Focus
Common Exam Questions
Clinical Scenarios:
- "A 68-year-old man presents with 18 months of progressive dyspnoea and dry cough. What are your differential diagnoses and how would you investigate?"
- "HRCT shows honeycombing with subpleural and basal predominance. What is the diagnosis and how would you manage this patient?"
- "What are the indications for surgical lung biopsy in suspected IPF?"
- "Compare and contrast IPF and chronic hypersensitivity pneumonitis."
- "A patient with IPF develops acute worsening of dyspnoea. What is your differential diagnosis and management approach?"
- "What is the role of antifibrotic therapy in IPF? Discuss the evidence."
- "When should a patient with IPF be referred for lung transplant assessment?"
Viva Points
Opening Statement:
"Idiopathic pulmonary fibrosis is a chronic, progressive, and irreversible fibrosing interstitial pneumonia of unknown cause, occurring primarily in older adults and characterised by the histopathological and radiological pattern of usual interstitial pneumonia. It has a median survival of 3-5 years and is the most common and most lethal form of idiopathic interstitial pneumonia."
Key Facts to Mention:
- Epidemiology: Incidence 3-9 per 100,000, median age 66-70 years, male predominance
- Pathophysiology: Unknown trigger leads to alveolar epithelial injury and aberrant wound healing with excessive fibrosis mediated by TGF-β and other pro-fibrotic pathways
- Clinical Presentation: Progressive dyspnoea, dry cough, inspiratory "Velcro" crackles (> 90%), clubbing (25-50%)
- Diagnosis: Requires exclusion of other ILDs and identification of UIP pattern on HRCT or biopsy; definite UIP pattern on HRCT in appropriate clinical context obviates need for biopsy (ATS/ERS/JRS/ALAT 2018)
- HRCT UIP Pattern: Subpleural and basal predominant honeycombing, reticular pattern, traction bronchiectasis, absence of features suggesting alternative diagnosis
- Differential Diagnosis: NSIP, chronic hypersensitivity pneumonitis, CTD-ILD, sarcoidosis, asbestosis
- Management: Antifibrotic therapy (pirfenidone or nintedanib) reduces FVC decline by ~50%; lung transplantation is only potentially curative treatment; supportive care includes oxygen, pulmonary rehabilitation, vaccination, symptom management
- Prognosis: Median survival 3-5 years; GAP index predicts mortality; acute exacerbations carry > 50% mortality
- Complications: Acute exacerbation (5-10% per year, > 50% mortality), pulmonary hypertension (30-50%), lung cancer (8-15%)
Common Mistakes
Diagnostic Errors:
❌ Failing to exclude alternative diagnoses: IPF is a diagnosis of exclusion. Always screen for CTD, obtain detailed exposure history, and exclude drug-induced ILD.
❌ Misinterpreting HRCT: Confusing UIP pattern with other patterns; missing features inconsistent with UIP (ground-glass predominance, upper lobe distribution, mosaic attenuation).
❌ Not recognising the need for MDD: Individual assessment is less accurate than multidisciplinary discussion.
Management Errors:
❌ Using corticosteroids or immunosuppression: These are NOT indicated for IPF and may cause harm.
❌ Delaying antifibrotic therapy: Should be initiated soon after diagnosis in eligible patients.
❌ Late referral for transplant: Patients should be referred early (at or shortly after diagnosis) as disease may progress rapidly and render them ineligible.
❌ Not recognising acute exacerbation: Failure to identify and manage this life-threatening complication.
Model Answers
Q: "A 65-year-old male ex-smoker presents with 12 months of progressive dyspnoea and dry cough. Examination reveals bibasal fine inspiratory crackles. How would you approach this patient?"
A: "This presentation is highly suggestive of interstitial lung disease, with IPF being a key differential given the patient's age, sex, smoking history, and clinical findings. My approach would be systematic:
History: I would obtain a detailed history including:
- Characterisation of dyspnoea and cough (duration, progression, exacerbating factors)
- Occupational and environmental exposure history (asbestos, birds, moulds, metal dusts)
- Medication history (drugs causing ILD: amiodarone, methotrexate, nitrofurantoin)
- Symptoms of connective tissue disease (arthritis, rashes, Raynaud's, dry eyes/mouth)
- Smoking history
Examination: I would perform a full respiratory examination looking for crackles (quality, distribution), clubbing, signs of right heart failure, and signs of connective tissue disease.
Investigations:
- HRCT chest: The key investigation to identify the pattern of ILD. In IPF, I would expect a UIP pattern with subpleural and basal predominant honeycombing, reticular pattern, and traction bronchiectasis without features inconsistent with UIP such as ground-glass predominance or upper lobe distribution.
- Pulmonary function tests: Typically show restrictive pattern (reduced FVC and TLC) with reduced DLCO disproportionate to lung volumes.
- Autoimmune screen: ANA, RF, anti-CCP, myositis panel to exclude CTD-ILD.
- 6-minute walk test: Assess exercise tolerance and oxygen desaturation.
Multidisciplinary Discussion: I would present the case at an ILD MDT involving respiratory physicians, radiologists, and pathologists to reach a consensus diagnosis.
Diagnosis: If HRCT shows definite UIP pattern in the appropriate clinical context with exclusion of alternative causes, the diagnosis of IPF can be made without surgical lung biopsy per ATS/ERS 2018 guidelines.
Management: For confirmed IPF, I would:
- Initiate antifibrotic therapy (pirfenidone or nintedanib) to slow disease progression
- Provide supplemental oxygen if hypoxaemic
- Refer for pulmonary rehabilitation
- Ensure vaccinations (influenza, pneumococcal, COVID-19)
- Refer for lung transplant assessment if eligible
- Discuss prognosis and advance care planning"
Q: "What is the difference between UIP pattern and probable UIP pattern on HRCT?"
A: "The ATS/ERS/JRS/ALAT 2018 guidelines define HRCT patterns with different levels of certainty for UIP:
UIP Pattern (Definite UIP) requires ALL of:
- Subpleural and basal predominance
- Honeycombing with or without peripheral traction bronchiectasis
- Reticular pattern
- Absence of features inconsistent with UIP
Probable UIP Pattern has:
- Subpleural and basal predominant reticular pattern
- Traction bronchiectasis or bronchiolectasis
- Absence of honeycombing (this is the key difference)
- Absence of features inconsistent with UIP
The distinction is clinically important because definite UIP pattern on HRCT in the appropriate clinical context is sufficient for IPF diagnosis without biopsy, whereas probable UIP pattern may warrant consideration of surgical lung biopsy depending on the clinical scenario and level of diagnostic confidence, though many patients with probable UIP pattern and typical clinical features can be diagnosed with IPF without biopsy."
References
-
Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68. doi:10.1164/rccm.201807-1255ST
-
Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018;378(19):1811-1823. doi:10.1056/NEJMra1705751
-
Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(4):431-440. doi:10.1164/rccm.201006-0894CI
-
Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J. 2012;40(3):519-521. doi:10.1183/09031936.00001612
-
King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083-2092. doi:10.1056/NEJMoa1402582
-
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082. doi:10.1056/NEJMoa1402584
-
Yusen RD, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report--2015. J Heart Lung Transplant. 2015;34(10):1264-1277. doi:10.1016/j.healun.2015.08.014
-
Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46(3):795-806. doi:10.1183/09031936.00185114
-
Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355-361. doi:10.1183/09059180.00002512
-
Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2(7):566-572. doi:10.1016/S2213-2600(14)70101-8
-
Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155(1):242-248. doi:10.1164/ajrccm.155.1.9001319
-
Tang YW, Johnson JE, Browning PJ, et al. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41(6):2633-2640. doi:10.1128/JCM.41.6.2633-2640.2003
-
Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317-1326. doi:10.1056/NEJMoa066157
-
Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503-1512. doi:10.1056/NEJMoa1013660
-
Lee JS, Ryu JH, Elicker BM, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(12):1390-1394. doi:10.1164/rccm.201101-0138OC
-
Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med. 2014;189(10):1161-1172. doi:10.1164/rccm.201312-2221PP
-
Königshoff M, Kramer M, Balsara N, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119(4):772-787. doi:10.1172/JCI33950
-
Schafer MJ, White TA, Iijima K, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi:10.1038/ncomms14532
-
Salisbury ML, Myers JL, Belloli EA, Kazerooni EA, Martinez FJ, Flaherty KR. Diagnosis and Treatment of Fibrotic Hypersensitivity Pneumonia. Where We Stand and Where We Need to Go. Am J Respir Crit Care Med. 2017;196(6):690-699. doi:10.1164/rccm.201608-1675PP
-
Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684-691. doi:10.7326/0003-4819-156-10-201205150-00004
-
Dowman L, Hill CJ, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst Rev. 2014;(10):CD006322. doi:10.1002/14651858.CD006322.pub3
-
Collard HR, Ryerson CJ, Corte TJ, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am J Respir Crit Care Med. 2016;194(3):265-275. doi:10.1164/rccm.201604-0801CI
Last Updated: 2026-01-05
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Differentials
Competing diagnoses and look-alikes to compare.
- Nonspecific Interstitial Pneumonia
- Chronic Hypersensitivity Pneumonitis
- Connective Tissue Disease-ILD
- Sarcoidosis
Consequences
Complications and downstream problems to keep in mind.
- Acute Respiratory Failure
- Pulmonary Hypertension