Systemic Mastocytosis
Clinical manifestations arise from two principal mechanisms: mast cell mediator release (causing flushing, urticaria, pruritus, anaphylaxis, gastrointestinal symptoms) and organ infiltration by neoplastic mast cells...
What matters first
Clinical manifestations arise from two principal mechanisms: mast cell mediator release (causing flushing, urticaria, pruritus, anaphylaxis, gastrointestinal symptoms) and organ infiltration by neoplastic mast cells...
Anaphylaxis
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Anaphylaxis
- Advanced systemic mastocytosis
- Organ damage (cytopenias, hepatosplenomegaly)
- Associated haematological neoplasm
Linked comparisons
Differentials and adjacent topics worth opening next.
- Anaphylaxis
- Urticaria
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Systemic Mastocytosis
1. Clinical Overview
Summary
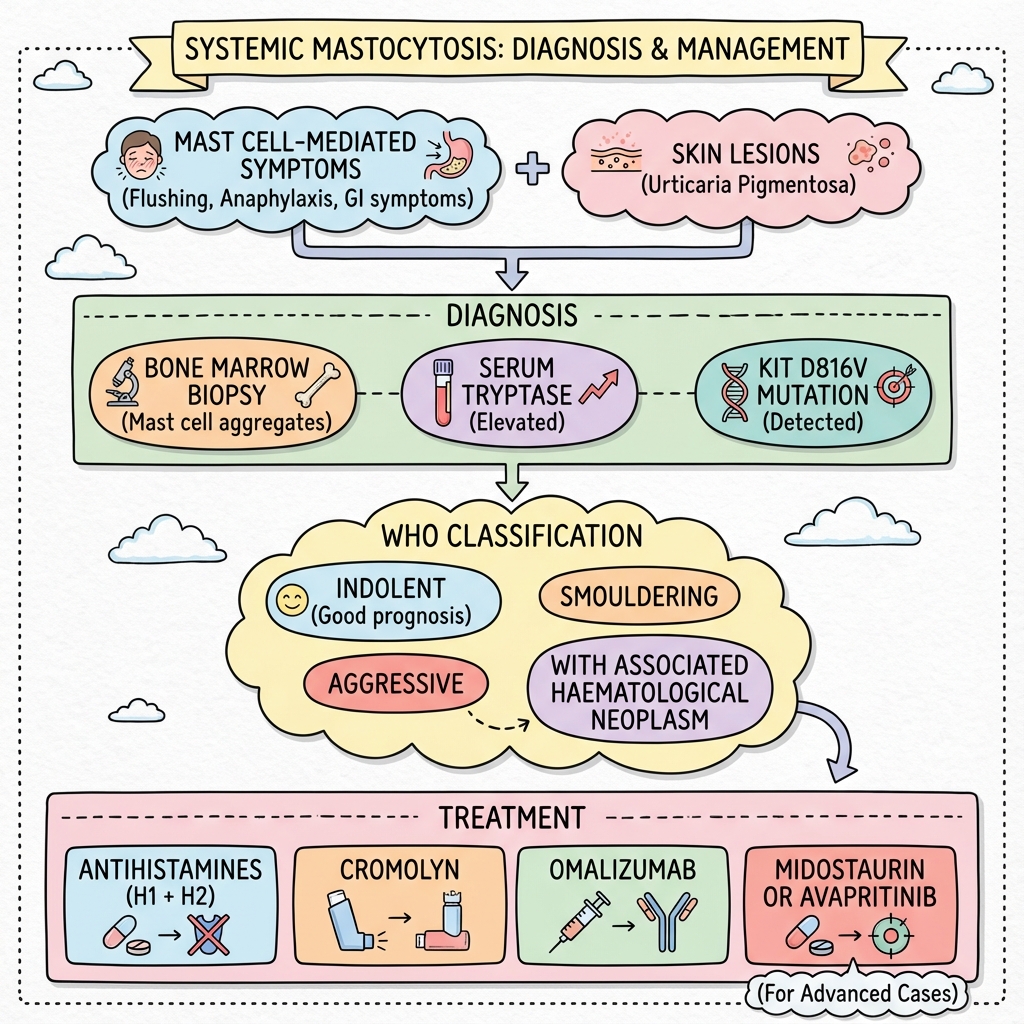
Systemic mastocytosis (SM) is a clonal haematological disorder characterised by abnormal proliferation and accumulation of neoplastic mast cells in one or more extracutaneous organs. The disease is classified as a myeloproliferative neoplasm in the 2016 WHO classification. [1] The vast majority of SM cases (approximately 90%) harbour an activating mutation in the KIT proto-oncogene, most commonly the D816V substitution, which results in constitutive activation of the KIT receptor tyrosine kinase and mast cell proliferation. [2]
Clinical manifestations arise from two principal mechanisms: mast cell mediator release (causing flushing, urticaria, pruritus, anaphylaxis, gastrointestinal symptoms) and organ infiltration by neoplastic mast cells (hepatosplenomegaly, lymphadenopathy, bone lesions, cytopenias). [3] Cutaneous involvement, presenting as urticaria pigmentosa with the pathognomonic Darier sign (wheal and flare upon rubbing), occurs in the majority of patients but is not universal. [4]
The WHO classification recognises several SM variants ranging from indolent SM (ISM) with normal life expectancy to aggressive SM (ASM), SM with associated haematological neoplasm (SM-AHN), and mast cell leukaemia (MCL) with poor prognosis. [1] Recent therapeutic advances include the KIT inhibitors midostaurin (FDA-approved 2017 for advanced SM) and avapritinib (approved 2021 for advanced SM), which have transformed outcomes in aggressive disease. [5,6]
Key Facts
- Definition: Clonal myeloproliferative neoplasm with abnormal mast cell accumulation in tissues
- Incidence: Rare disease, estimated 0.5-1 cases per 100,000 per year [7]
- Peak Demographics: Adults (median age 50-60 years); slight male predominance
- Pathognomonic: KIT D816V mutation (90% of SM) + serum tryptase > 20 ng/mL + multifocal mast cell aggregates in bone marrow
- Gold Standard Investigation: Bone marrow biopsy with immunohistochemistry (CD117, CD25, CD2) + KIT mutation analysis + serum tryptase
- First-line Treatment: H1 and H2 antihistamines for symptom control; epinephrine autoinjector; targeted therapy (midostaurin/avapritinib) for advanced disease
- Prognosis: ISM - normal to near-normal life expectancy; ASM - median survival 3-5 years untreated; MCL - median survival less than 6 months [8]
Clinical Pearls
Diagnostic Pearl: Serum tryptase persistently > 20 ng/mL is a major criterion for SM diagnosis. A level > 200 ng/mL suggests advanced disease and correlates with higher disease burden. [9]
Emergency Pearl: Patients with mastocytosis are at high risk for severe, potentially fatal anaphylaxis triggered by Hymenoptera stings, anaesthetic agents, radiocontrast, NSAIDs, and antibiotics. All patients should carry two epinephrine autoinjectors. [10]
Treatment Pearl: The D816V mutation confers resistance to imatinib but sensitivity to midostaurin and avapritinib. Always perform KIT mutation analysis before initiating targeted therapy. [5,6]
Examination Pearl: Darier sign (wheal and flare upon gentle rubbing or stroking of skin lesions) is pathognomonic for cutaneous mastocytosis and present in 70-90% of patients with skin involvement. [4]
2. Epidemiology
Systemic mastocytosis is a rare disease with true incidence difficult to establish due to underdiagnosis and variable presentation. Population-based studies estimate an annual incidence of 0.5-1.0 cases per 100,000 population in Western countries. [7] The prevalence is estimated at 1 in 10,000 to 1 in 20,000 individuals. [11]
| Epidemiological Parameter | Value | Source |
|---|---|---|
| Annual incidence | 0.5-1.0 per 100,000 | [7] |
| Estimated prevalence | 1 in 10,000-20,000 | [11] |
| Median age at diagnosis | 50-60 years | [12] |
| Male:Female ratio | 1.2-1.5:1 | [12] |
| Proportion with KIT D816V | 85-95% | [2] |
| Proportion with cutaneous involvement | 70-90% | [4] |
| ISM proportion of all SM | 70-80% | [1] |
| ASM/SM-AHN/MCL proportion | 20-30% | [1] |
Demographics and Risk Factors
Systemic mastocytosis predominantly affects adults, with a median age at diagnosis of 50-60 years. [12] There is a slight male predominance (male-to-female ratio approximately 1.2-1.5:1). [12] Unlike cutaneous mastocytosis in children, which often resolves spontaneously, adult-onset disease is typically persistent and systemic.
The vast majority of cases are sporadic with no identified environmental risk factors. Familial clustering is rare (less than 5% of cases), and most familial cases are associated with germline KIT mutations. [13] There are no established associations with ethnicity, occupation, or geographic location.
Disease Subtypes Distribution
Among patients diagnosed with systemic mastocytosis, indolent SM (ISM) accounts for 70-80% of cases and has an excellent prognosis with normal or near-normal life expectancy. [1,8] Smouldering SM (SSM), a variant of ISM with higher disease burden but no organ damage, represents 5-10% of cases. [1]
Advanced SM variants, including aggressive SM (ASM), SM with associated haematological neoplasm (SM-AHN), and mast cell leukaemia (MCL), collectively comprise 20-30% of cases. [1] SM-AHN is the most common advanced variant, with the associated haematological neoplasm most frequently being a myeloid disorder (acute myeloid leukaemia, myelodysplastic syndrome, myeloproliferative neoplasm). [14]
3. Aetiology and Pathophysiology
Molecular Genetics
The hallmark of systemic mastocytosis is the presence of activating mutations in the KIT proto-oncogene (chromosome 4q12), which encodes the receptor tyrosine kinase for stem cell factor (SCF). [2] The D816V mutation, a substitution of aspartate with valine at codon 816 in the kinase domain, is detected in 85-95% of adult SM patients. [2] This mutation results in constitutive, ligand-independent activation of KIT signalling, driving mast cell proliferation, survival, and resistance to apoptosis.
The D816V mutation confers resistance to imatinib (which binds the inactive conformation of KIT) but sensitivity to Type II KIT inhibitors that target the active conformation, such as midostaurin and avapritinib. [5,6] Rare KIT mutations include D816Y, D816F, D816H, and mutations at other codons (V560G, deletion of codon 419). [15]
Additional somatic mutations are frequently identified in advanced SM, particularly in patients with SM-AHN. These include mutations in TET2 (20-30%), SRSF2 (15-25%), ASXL1 (10-20%), RUNX1 (10-15%), and CBL (5-10%), which contribute to disease progression and adverse prognosis. [14,16]
Pathophysiology
The pathophysiology of systemic mastocytosis involves both the proliferative expansion of neoplastic mast cells and the release of vasoactive and inflammatory mediators.
Mast Cell Proliferation and Accumulation: Constitutive activation of KIT signalling through the D816V mutation activates multiple downstream pathways:
- RAS/RAF/MEK/ERK pathway: Promotes cell proliferation and survival
- PI3K/AKT pathway: Inhibits apoptosis and enhances cell survival
- JAK/STAT pathway: Drives proliferation and cytokine production
- SRC family kinases: Mediate cell adhesion and migration
The neoplastic mast cells accumulate in various tissues, forming multifocal aggregates particularly in bone marrow, skin, gastrointestinal tract, liver, spleen, and lymph nodes. [3]
Aberrant Mast Cell Phenotype: Neoplastic mast cells exhibit abnormal immunophenotype, expressing:
- CD117 (KIT) - universal marker
- CD25 (IL-2 receptor alpha chain) - aberrant expression, diagnostic marker
- CD2 (T-cell marker) - aberrant co-expression in most cases
- Spindle-shaped morphology (in contrast to normal round mast cells) [1]
Mediator Release: Both neoplastic and residual normal mast cells release numerous preformed and newly synthesised mediators:
Preformed mediators:
- Histamine: Vasodilation, increased vascular permeability, pruritus, gastric acid secretion
- Tryptase: Serum marker; promotes fibrinolysis and inflammation
- Heparin: Anticoagulation (rarely clinically significant)
Newly synthesised mediators:
- Prostaglandin D2: Vasodilation, bronchoconstriction, GI hypermotility
- Leukotrienes (C4, D4, E4): Bronchoconstriction, increased vascular permeability
- Platelet-activating factor: Hypotension, bronchoconstriction
Cytokines:
- IL-6, IL-13, TNF-alpha: Systemic inflammation, constitutional symptoms
The combination of mediator release and organ infiltration results in the diverse clinical manifestations of SM. [3,10]
Disease Progression
Indolent SM typically remains stable for years to decades, with only 5-10% of patients progressing to advanced disease over 20 years. [8] Factors associated with progression include high baseline tryptase (> 200 ng/mL), lack of skin involvement, presence of B and/or C findings, and acquisition of additional mutations (TET2, SRSF2, ASXL1). [16]
4. Clinical Presentation
The clinical manifestations of systemic mastocytosis result from mast cell mediator release and/or organ infiltration. Presentation is highly variable, ranging from minimal symptoms in ISM to multiorgan failure in advanced disease.
Symptoms Related to Mediator Release
Constitutional Symptoms
- Fatigue and malaise: Present in 50-70% of patients [3]
- Headache: Often severe, may be migraine-like
- Cognitive difficulties: "Brain fog," impaired concentration
Cutaneous Symptoms
- Flushing: Spontaneous or triggered (70% of patients) [10]
- Pruritus: Generalised or localised to skin lesions (60-80%)
- Urticaria: Recurrent episodes independent of skin lesions
Gastrointestinal Symptoms
- Abdominal pain and cramping: Episodic or chronic (60-80%) [17]
- Diarrhoea: Often post-prandial, may be severe
- Nausea and vomiting: Triggered episodes
- Gastroesophageal reflux: Due to increased gastric acid secretion
- Malabsorption: Rare, in severe cases
Cardiovascular Symptoms
- Hypotensive episodes: Presyncope or syncope (20-40%)
- Tachycardia: Associated with flushing or anaphylaxis
- Chest discomfort: May mimic angina
Anaphylaxis
- Severe, life-threatening episodes: 20-50% of SM patients experience at least one episode [10]
- Common triggers: Hymenoptera stings (especially wasps/bees), general anaesthesia, radiocontrast media, NSAIDs, opioids, antibiotics, alcohol
- Atypical presentation: May lack urticaria; profound hypotension predominates
Signs Related to Organ Infiltration
Cutaneous Manifestations
- Urticaria pigmentosa: Brown to reddish-brown macules or papules (70-90% of patients with skin involvement) [4]
- Darier sign: Pathognomonic; wheal and flare upon gentle stroking of lesions (present within 2-5 minutes)
- Distribution: Trunk and proximal extremities most common; usually spares face, palms, soles
- Telangiectasia macularis eruptiva perstans: Variant with telangiectatic macules
Hepatosplenic Involvement
- Hepatomegaly: 40-60% in advanced SM [1]
- Splenomegaly: 50-70% in advanced SM
- Portal hypertension: Rare, due to mast cell infiltration or fibrosis
- Ascites: In advanced cases with portal hypertension
Lymphadenopathy
- Generalised or localised: 20-40% in advanced SM
- Non-tender, firm nodes: Mast cell infiltration
Skeletal Manifestations
- Bone pain: 20-30% of patients, particularly axial skeleton [18]
- Osteoporosis/osteopenia: 30-50% of patients; increased fracture risk
- Osteosclerosis: Less common than osteopenia
- Pathological fractures: Vertebral compression fractures most common
Haematological Manifestations (Advanced SM)
- Anaemia: Due to bone marrow infiltration or associated haematological neoplasm
- Thrombocytopenia: In advanced disease
- Leucopenia or leucocytosis: Depending on subtype and associated neoplasm
- Eosinophilia: Occasionally present
| Clinical Feature | Frequency in SM | Clinical Significance |
|---|---|---|
| Urticaria pigmentosa | 70-90% | Highly suggestive; Darier sign pathognomonic |
| Flushing | 60-80% | Mediator release; distinguish from carcinoid |
| Gastrointestinal symptoms | 60-80% | Major impact on quality of life |
| Anaphylaxis | 20-50% | Life-threatening; requires epinephrine |
| Hepatosplenomegaly | 40-70% in ASM | Indicates organ infiltration; C-finding |
| Bone pain | 20-30% | Often due to osteoporosis or infiltration |
| Constitutional symptoms | 50-70% | Fatigue, malaise, weight loss |
5. Differential Diagnosis
The differential diagnosis of systemic mastocytosis depends on the predominant clinical presentation.
For Patients Presenting with Flushing and Mediator-Release Symptoms
-
Carcinoid Syndrome (Most common must-not-miss)
- Key distinguishing features: 5-HIAA elevated in urine; hepatic metastases on imaging; diarrhoea more prominent; tryptase normal; skin lesions absent
- Investigations: 24-hour urinary 5-HIAA, chromogranin A, somatostatin receptor imaging
-
Phaeochromocytoma
- Key distinguishing features: Episodic hypertension (not hypotension); paroxysmal headache, sweating, palpitations; elevated metanephrines; no skin lesions
- Investigations: Plasma or urinary metanephrines, adrenal imaging
-
Hereditary Alpha-Tryptasemia (HαT)
- Key distinguishing features: Familial; baseline tryptase elevated but typically less than 20 ng/mL; no bone marrow aggregates; no organomegaly
- Investigations: TPSAB1 gene copy number; normal bone marrow
-
Anaphylaxis (Idiopathic or Known Trigger)
- Key distinguishing features: Episodic only (not chronic symptoms); tryptase may be transiently elevated during episodes but normal baseline; no skin lesions or organomegaly
- Investigations: Baseline tryptase (if elevated, investigate for SM)
-
Menopause/Hormonal Flushing
- Key distinguishing features: Perimenopausal women; hot flushes without urticaria or hypotension; no GI symptoms; normal tryptase
- Investigations: FSH/LH, tryptase
For Patients Presenting with Urticarial Skin Lesions
-
Chronic Urticaria
- Key distinguishing features: Wheals are transient (resolve less than 24 hours); no fixed pigmented lesions; negative Darier sign; normal tryptase
- Investigations: Urticaria activity score; tryptase
-
Urticarial Vasculitis
- Key distinguishing features: Lesions painful rather than pruritic; last > 24 hours; may leave hyperpigmentation; normal tryptase; skin biopsy shows vasculitis
- Investigations: Complement levels, ANA, skin biopsy
For Patients Presenting with Haematological Abnormalities
-
Other Myeloproliferative Neoplasms (CML, PV, ET, PMF)
- Key distinguishing features: Different driver mutations (BCR-ABL1, JAK2 V617F, CALR, MPL); different blood count patterns; no skin lesions or anaphylaxis
- Investigations: BCR-ABL1, JAK2 mutation, bone marrow biopsy
-
Myelodysplastic Syndrome
- Key distinguishing features: Cytopenias; dysplasia on bone marrow; no skin lesions or mediator symptoms; normal tryptase
- Investigations: Bone marrow morphology, cytogenetics
| Differential | Key Distinguishing Features | Critical Investigation |
|---|---|---|
| Carcinoid syndrome | 5-HIAA elevated; hepatic metastases; normal tryptase | Urinary 5-HIAA |
| Phaeochromocytoma | Episodic hypertension; elevated metanephrines | Plasma metanephrines |
| Hereditary alpha-tryptasemia | Familial; tryptase less than 20 ng/mL; normal marrow | TPSAB1 copy number |
| Chronic urticaria | Transient wheals; no pigmentation; normal tryptase | Baseline tryptase |
| CML | BCR-ABL1 positive; high WCC; no skin/mediator symptoms | BCR-ABL1 PCR |
6. Investigations
The diagnosis of systemic mastocytosis requires demonstration of pathological mast cell accumulation in extracutaneous tissues, typically bone marrow, according to WHO criteria.
First-Line Investigations
Serum Tryptase
- Baseline level: Most important initial screening test
- Interpretation:
- "less than 11.4 ng/mL: Normal; SM very unlikely"
- "11.4-20 ng/mL: Borderline; consider hereditary alpha-tryptasemia or repeat testing"
- "> 20 ng/mL: Major criterion for SM; warrants bone marrow biopsy [1,9]"
- "> 200 ng/mL: Suggests advanced SM; high disease burden [9]"
- Timing: Must be measured at baseline (not during acute event)
- Pearls: Elevated tryptase alone is not diagnostic; requires bone marrow confirmation. Hereditary alpha-tryptasemia can cause baseline elevation less than 20 ng/mL
Complete Blood Count with Differential
- Purpose: Detect cytopenias (C-finding) or associated haematological neoplasm
- Findings: Usually normal in ISM; anaemia, thrombocytopenia, leucopenia, or eosinophilia may be present in advanced SM
Liver and Renal Function Tests
- Purpose: Assess for organ dysfunction (C-findings)
- Findings: Elevated alkaline phosphatase or transaminases in hepatic involvement
Bone Density Scan (DEXA)
- Purpose: Screen for osteoporosis (30-50% of patients) [18]
- Timing: At diagnosis and periodically
Definitive Diagnostic Investigations
Bone Marrow Biopsy and Aspirate
- Gold standard for SM diagnosis
- Procedure: Typically from posterior iliac crest
- Histology:
- "Multifocal mast cell aggregates (≥15 mast cells per aggregate): Major criterion [1]"
- Spindle-shaped mast cells
- Mast cell infiltration often paratrabecular
- Immunohistochemistry:
- "CD117 (KIT): Positive (universal)"
- "CD25: Aberrant expression (minor criterion) [1]"
- "CD2: Aberrant co-expression (minor criterion) [1]"
- "Tryptase staining: Highlights mast cells"
- Flow cytometry: Demonstrates aberrant CD25 and/or CD2 expression
- Morphology: ≥25% spindle-shaped or atypical mast cells (minor criterion) [1]
KIT Mutation Analysis
- Method: PCR-based techniques; highly sensitive allele-specific PCR or next-generation sequencing
- Sample: Bone marrow aspirate, peripheral blood (less sensitive), or skin biopsy
- Findings:
- D816V mutation detected in 85-95% of SM cases (minor criterion) [1,2]
- Other activating KIT mutations in remaining cases
- Clinical significance: Predicts response to therapy; D816V confers imatinib resistance
WHO Diagnostic Criteria for Systemic Mastocytosis
Major Criterion (1):
- Multifocal dense aggregates of mast cells (≥15 mast cells per aggregate) in bone marrow or other extracutaneous tissues
Minor Criteria (4):
-
25% of mast cells in bone marrow or extracutaneous tissues are spindle-shaped or atypical morphology
- Detection of KIT codon 816 mutation (or other activating mutation) in bone marrow or extracutaneous organ
- Aberrant expression of CD25 and/or CD2 on mast cells in bone marrow or extracutaneous tissues
- Serum total tryptase persistently > 20 ng/mL (not valid if associated clonal myeloid disorder present)
Diagnosis requires:
- 1 major + 1 minor criterion, OR
- 3 minor criteria [1]
Second-Line and Specialist Investigations
Additional Mutation Analysis
- Panel testing: TET2, SRSF2, ASXL1, RUNX1, CBL mutations
- Purpose: Prognostic stratification; often present in SM-AHN [14,16]
Imaging Studies
Skeletal Imaging:
- Plain radiographs: Detect osteolytic or osteosclerotic lesions
- CT or MRI: Better assessment of bone involvement
- Findings: Osteopenia/osteoporosis, focal sclerotic or lytic lesions (20-30% of patients)
Abdominal Imaging:
- Ultrasound or CT: Assess hepatosplenomegaly, lymphadenopathy
- Purpose: Identify organomegaly (C-finding in advanced SM)
Gastrointestinal Endoscopy
- Indications: Severe GI symptoms; suspected GI infiltration
- Findings: Normal mucosa or non-specific inflammation; biopsy may show mast cell infiltration
Risk Stratification: REMA Score
The Response to Midostaurin, Anaphylaxis (REMA) score is a validated tool for anaphylaxis risk assessment in mastocytosis patients: [10]
REMA Score Components:
- Recurrent anaphylaxis
- Elevated tryptase
- Male sex
- Absence of skin involvement
Higher REMA scores correlate with increased anaphylaxis risk.
| Investigation | Finding in SM | Sensitivity | Specificity |
|---|---|---|---|
| Serum tryptase > 20 ng/mL | Major criterion | 85-90% | 80-90% |
| Bone marrow mast cell aggregates | Major criterion | 95-100% | > 95% |
| KIT D816V mutation | Minor criterion | 85-95% | > 95% |
| CD25 expression on mast cells | Minor criterion | 90-95% | > 90% |
| CD2 expression on mast cells | Minor criterion | 70-80% | > 90% |
7. WHO Classification and Staging
The 2016 WHO classification divides systemic mastocytosis into distinct variants based on disease burden, organ involvement, and prognosis. [1]
Cutaneous Mastocytosis (CM)
Mast cell infiltration limited to skin; no evidence of systemic involvement.
- Does not meet criteria for SM
- Includes urticaria pigmentosa, diffuse cutaneous mastocytosis, mastocytoma
- More common in children; often resolves spontaneously
Systemic Mastocytosis Variants
1. Indolent Systemic Mastocytosis (ISM)
Definition: Meets SM criteria; no C-findings (organ damage); no associated haematological neoplasm
Characteristics:
- Most common variant (70-80% of SM)
- Excellent prognosis; normal to near-normal life expectancy [8]
- Low disease burden; predominantly mediator symptoms
- Skin involvement common
B-findings (high disease burden, but no organ damage):
-
30% mast cell infiltration in bone marrow biopsy
- Serum tryptase > 200 ng/mL
- Signs of dysplasia or myeloproliferation without criteria for associated haematological neoplasm
- Hepatomegaly, splenomegaly, or lymphadenopathy without organ dysfunction
Bone Marrow Mastocytosis (BMM): Subvariant of ISM with bone marrow involvement but no skin lesions
Smouldering Systemic Mastocytosis (SSM): ISM with ≥2 B-findings; higher risk of progression but still good prognosis
2. Systemic Mastocytosis with Associated Haematological Neoplasm (SM-AHN)
Definition: Meets SM criteria + concurrent WHO-defined haematological neoplasm
Characteristics:
- 20-30% of SM cases [14]
- Associated neoplasm most commonly:
- Acute myeloid leukaemia (AML)
- Myelodysplastic syndrome (MDS)
- Myeloproliferative neoplasm (MPN)
- Chronic myelomonocytic leukaemia (CMML)
- "Less commonly: lymphoid malignancies"
- Prognosis determined by both SM variant and associated neoplasm
- Often harbours additional mutations (TET2, SRSF2, ASXL1) [16]
3. Aggressive Systemic Mastocytosis (ASM)
Definition: Meets SM criteria + ≥1 C-finding (organ damage due to mast cell infiltration); no associated haematological neoplasm
C-findings (organ damage):
- Cytopenia(s): ANC less than 1.0 × 10⁹/L, Hb less than 10 g/dL, or platelets less than 100 × 10⁹/L
- Hepatomegaly with impaired liver function, ascites, or portal hypertension
- Skeletal involvement with large osteolytic lesions and/or pathological fractures
- Splenomegaly with hypersplenism
- Malabsorption with weight loss due to GI mast cell infiltration
Characteristics:
- 5-10% of SM cases
- Median survival 3-5 years without treatment [8]
- Rapid progression; requires cytoreductive therapy
- Less commonly has skin involvement
4. Mast Cell Leukaemia (MCL)
Definition: Mast cells ≥10% of peripheral blood white cells and/or ≥20% in bone marrow aspirate
Variants:
- Classical MCL: Meets criteria above; rare (less than 1% of SM); median survival less than 6 months [8]
- Aleukemic MCL: less than 10% mast cells in blood but ≥20% in bone marrow; slightly better prognosis
Characteristics:
- Extremely poor prognosis
- Multi-organ failure common
- Skin involvement rare
- Requires urgent intensive therapy
| WHO Subtype | Frequency | Organ Damage | Associated Neoplasm | Median Survival |
|---|---|---|---|---|
| ISM | 70-80% | No (no C-findings) | No | Normal lifespan |
| SSM | 5-10% | No (but ≥2 B-findings) | No | Decades |
| SM-AHN | 20-30% | Variable | Yes | Variable (depends on AHN) |
| ASM | 5-10% | Yes (≥1 C-finding) | No | 3-5 years untreated |
| MCL | less than 1% | Severe | No | less than 6 months |
Prognostic Factors
Favourable:
- Indolent variant
- Skin involvement (urticaria pigmentosa)
- Younger age
- KIT D816V mutation alone (no additional mutations)
- Serum tryptase less than 200 ng/mL
Unfavourable:
- Advanced SM (ASM, MCL)
- Absence of skin involvement
- C-findings present
- Additional mutations (TET2, SRSF2, ASXL1, RUNX1) [16]
- Serum tryptase > 200 ng/mL
- Thrombocytopenia
8. Management
Management is stratified by disease variant and focuses on symptom control (ISM) versus cytoreduction and targeted therapy (advanced SM).
Management Algorithm
SM Diagnosis Confirmed (WHO criteria)
|
v
Classify Variant (ISM vs Advanced)
|
+----+----+
| |
v v
ISM Advanced SM (ASM/SM-AHN/MCL)
| |
v v
Symptom Assess for
Control Cytoreductive
Therapy
| |
v v
H1+H2 KIT Mutation
Anti- Status
histamines |
| +----+----+
| | |
v v v
Mast Cell D816V+ D816V-
Stabilizers |
| | v
v v Imatinib
Epinephrine Midostaurin or
Auto- or Cladribine
injector Avapritinib
| |
v v
Monitoring Monitor Response
Consider SCT
General Measures for All Patients
Trigger Avoidance
- Hymenoptera sting avoidance: Protective clothing; avoid provocative scents/foods outdoors
- Medications to avoid/use with caution:
- NSAIDs (especially aspirin) - use COX-2 selective if needed
- Opioids (especially codeine, morphine) - prefer oxycodone or fentanyl if needed
- Radiocontrast media - premedicate with antihistamines and corticosteroids
- Anaesthetic agents - inform anaesthetist; avoid direct mast cell degranulators
- Vancomycin - infuse slowly to avoid "red man syndrome"
- Physical triggers: Avoid extreme temperature changes, vigorous rubbing of skin, emotional stress
- Alcohol: May trigger flushing and mediator release
Emergency Preparedness
- Epinephrine autoinjectors: All patients should carry two devices (repeat dose may be needed) [10]
- Medical alert bracelet: Identify mastocytosis and anaphylaxis risk
- Anaphylaxis action plan: Written instructions for patient and family
Management of Indolent SM (ISM/SSM)
Goal: Symptom control; cytoreduction not required.
First-Line Pharmacotherapy
H1 Antihistamines:
- Non-sedating: Cetirizine 10-20 mg daily, loratadine 10 mg daily, fexofenadine 180 mg daily
- Sedating (if needed for sleep/pruritus): Diphenhydramine 25-50 mg nightly, hydroxyzine 25-50 mg nightly
- Dosing: Often require higher than standard doses (up to 4× licensed dose) for adequate symptom control [17]
H2 Antihistamines:
- Ranitidine: 150-300 mg twice daily (note: withdrawn in some countries due to contamination concerns)
- Famotidine: 20-40 mg twice daily
- Purpose: Reduce gastric acid secretion; control GI symptoms
Mast Cell Stabilizers:
- Cromolyn sodium (sodium cromoglicate): 200 mg orally four times daily
- Indication: Gastrointestinal symptoms (diarrhoea, abdominal pain) refractory to antihistamines [17]
- Mechanism: Inhibits mast cell degranulation; poorly absorbed (acts locally in GI tract)
Second-Line Pharmacotherapy
Leukotriene Receptor Antagonists:
- Montelukast: 10 mg daily
- Indication: Flushing, pruritus, respiratory symptoms refractory to antihistamines
Proton Pump Inhibitors:
- Omeprazole, lansoprazole, esomeprazole: Standard doses
- Indication: Refractory gastroesophageal reflux, peptic ulcer disease
Corticosteroids:
- Indication: Severe malabsorption, ascites (rare)
- Use with caution: Increased osteoporosis risk; reserve for severe cases
Antidepressants (SSRIs/SNRIs):
- Indication: Concurrent depression/anxiety common in chronic disease; may also help pain
Bisphosphonates:
- Indication: Osteoporosis (present in 30-50% of patients) [18]
- Options: Alendronate, risedronate, zoledronic acid
- Monitoring: Serial DEXA scans
Refractory Mediator Symptoms
Omalizumab (Anti-IgE Monoclonal Antibody):
- Dosing: 150-300 mg subcutaneously every 2-4 weeks
- Indication: Recurrent anaphylaxis despite antihistamines; refractory urticaria [19]
- Evidence: Case series and small studies show benefit in reducing anaphylaxis frequency
Interferon-alpha:
- Rarely used due to toxicity; reserved for severe refractory cases
Management of Advanced SM (ASM/SM-AHN/MCL)
Goal: Cytoreduction to reduce organ damage and improve survival.
KIT Inhibitor Therapy
Midostaurin (Type II Multi-Kinase Inhibitor):
- Approval: FDA-approved 2017 for advanced SM (ASM, SM-AHN, MCL) [5]
- Dosing: 100 mg orally twice daily (continuous)
- Mechanism: Inhibits KIT D816V, as well as FLT3, PDGFR, PKC
- Efficacy: Phase II trial showed 60% overall response rate in advanced SM; median duration of response 24 months [5]
- Side effects: Nausea/vomiting (most common; often improves with time), fatigue, cytopenias, elevated liver enzymes
- Monitoring: CBC, LFTs weekly for 4 weeks, then every 2 weeks for 8 weeks, then monthly
Avapritinib (Highly Selective KIT D816V Inhibitor):
- Approval: FDA-approved 2021 for advanced SM [6]
- Dosing: Starting dose 200 mg orally daily; may reduce to 100 mg or 50 mg for toxicity
- Mechanism: Type I KIT inhibitor; highly selective for KIT D816V mutation
- Efficacy: Phase I EXPLORER trial showed 75% overall response rate in advanced SM; superior to midostaurin in head-to-head comparison in PATHFINDER trial [6,20]
- Side effects: Cognitive effects (40-50%; "brain fog," memory impairment, mood changes - usually resolve with dose reduction), peripheral oedema, nausea, thrombocytopenia, anaemia
- Advantages: More selective; potentially more effective than midostaurin
- Monitoring: Neuropsychiatric assessment; CBC, LFTs regularly
Choice of KIT Inhibitor:
- Avapritinib generally preferred for D816V+ advanced SM due to higher response rates [20]
- Midostaurin alternative if avapritinib not available or not tolerated
- Both agents target D816V; imatinib is NOT effective
For D816V-Negative SM (Rare):
- Imatinib: 400 mg daily may be effective for wild-type KIT or certain non-D816V mutations (F522C, K509I, V560G)
- Cladribine: Cytotoxic agent; 0.13 mg/kg/day IV for 5 days every 4-8 weeks
Management of SM-AHN
Requires treatment of both the SM component and the associated haematological neoplasm:
- SM component: KIT inhibitor (midostaurin or avapritinib) as above
- AHN component: Appropriate therapy for the specific neoplasm (e.g., cytarabine-based chemotherapy for AML, hypomethylating agents for MDS, hydroxyurea or ruxolitinib for MPN)
- Sequencing: Simultaneous treatment of both components often necessary; coordinate with haematology expertise
Haematopoietic Stem Cell Transplantation (HSCT)
- Indication: Selected patients with advanced SM, particularly younger patients with ASM or MCL who achieve remission or disease control with KIT inhibitors
- Type: Allogeneic HSCT (curative potential)
- Evidence: Limited data; small case series suggest potential for cure, but high transplant-related mortality [21]
- Timing: Consider after initial disease control with targeted therapy
Cytoreductive Therapy (Alternative Agents)
Cladribine (2-CdA):
- Dosing: 0.13 mg/kg/day IV for 5 days every 4-8 weeks
- Efficacy: 50% response rate in advanced SM in small studies
- Toxicity: Myelosuppression, infections
Interferon-alpha:
- Historical agent; rarely used due to toxicity and availability of better options
Special Considerations
Perioperative Management
- Premedication: H1 and H2 antihistamines, corticosteroids (e.g., prednisolone 30-50 mg night before and morning of surgery)
- Anaesthetic agents: Avoid known mast cell degranulators (e.g., atracurium, mivacurium, morphine); prefer rocuronium, vecuronium, fentanyl, remifentanil
- Monitoring: Close monitoring for hypotension/anaphylaxis; have epinephrine readily available
- Inform anaesthetist: Ensure awareness of diagnosis and risk
Pregnancy
- Generally well tolerated: Pregnancy does not worsen SM in most cases
- Medications: Continue H1/H2 antihistamines (most are safe); avoid cromolyn in first trimester; avoid KIT inhibitors (teratogenic)
- Delivery: Vaginal delivery preferred; epidural anaesthesia generally safe; have epinephrine available; premedicate as for surgery
Anaphylaxis Management
- Immediate: Epinephrine 0.3-0.5 mg IM (1:1000) into anterolateral thigh; repeat every 5-15 minutes if needed
- Supportive: IV fluids, oxygen, supine position with legs elevated
- Second-line: H1 antihistamine (diphenhydramine 25-50 mg IV), H2 antihistamine (ranitidine 50 mg IV or famotidine 20 mg IV), corticosteroids (hydrocortisone 200 mg IV or methylprednisolone 125 mg IV)
- Observation: Minimum 4-6 hours (biphasic reactions possible); 12-24 hours if severe
- Hymenoptera venom immunotherapy: Recommended for patients with anaphylaxis to Hymenoptera stings; highly effective in reducing recurrence risk [10]
| Treatment | Indication | Mechanism | Evidence Level |
|---|---|---|---|
| H1 + H2 antihistamines | All patients; mediator symptoms | Block histamine receptors | High [17] |
| Cromolyn sodium | GI symptoms | Mast cell stabilization | Moderate [17] |
| Epinephrine autoinjector | All patients; anaphylaxis risk | Emergency treatment | High [10] |
| Omalizumab | Refractory anaphylaxis | Anti-IgE antibody | Moderate [19] |
| Midostaurin | Advanced SM (D816V+) | KIT inhibitor | High [5] |
| Avapritinib | Advanced SM (D816V+) | Selective KIT D816V inhibitor | High [6,20] |
| Imatinib | D816V-negative SM | KIT inhibitor (wild-type) | Moderate |
| Bisphosphonates | Osteoporosis | Bone resorption inhibition | High [18] |
Monitoring and Follow-Up
ISM:
- Clinical review: Every 6-12 months
- Serum tryptase: Annually (or if symptoms worsen)
- CBC, LFTs: Annually
- DEXA scan: Every 2-3 years or if osteoporosis detected
Advanced SM on Targeted Therapy:
- Clinical review: Every 4-8 weeks initially, then every 3 months
- Serum tryptase: Every 3 months (marker of disease burden)
- CBC, LFTs: Every 2-4 weeks initially (monitor for toxicity), then monthly
- Bone marrow biopsy: Every 6-12 months (assess response)
- Imaging (CT/ultrasound): Every 6 months (assess organomegaly)
9. Complications
| Complication | Frequency | Prevention | Management |
|---|---|---|---|
| Anaphylaxis | 20-50% lifetime [10] | Trigger avoidance; carry epinephrine | Epinephrine IM; IV fluids; antihistamines; corticosteroids |
| Osteoporosis/fractures | 30-50% [18] | Calcium/vitamin D; DEXA screening | Bisphosphonates; vertebroplasty for fractures |
| Peptic ulcer disease | 10-20% | H2 antagonists or PPIs | PPIs; eradicate H. pylori |
| Malabsorption | 5-10% in ISM; higher in ASM | Cromolyn; symptom control | Nutritional support; corticosteroids if severe |
| Hepatic dysfunction | Rare in ISM; 40-60% in ASM | Early treatment of advanced disease | KIT inhibitors; monitor liver function |
| Cytopenias | Rare in ISM; common in ASM/MCL | Early cytoreductive therapy | KIT inhibitors; transfusion support; G-CSF |
| Progression to advanced SM | 5-10% of ISM over 20 years [8] | Regular monitoring | Initiate KIT inhibitor therapy |
| Transformation to mast cell leukaemia | 1-2% | Early treatment; unclear if preventable | Urgent KIT inhibitor; consider HSCT |
| Depression/anxiety | 30-40% | Psychological support; patient education | SSRIs/SNRIs; cognitive behavioural therapy |
| Cardiovascular events | Increased risk due to mediator release | Control mediator symptoms | Standard cardiology management |
Anaphylaxis
The most feared complication, occurring in 20-50% of mastocytosis patients during their lifetime. [10] Risk factors include male sex, elevated tryptase, recurrent previous anaphylaxis, absence of skin involvement (captured in the REMA score). Triggers include Hymenoptera stings (most common), medications, and foods. Presentation may be atypical with profound hypotension and minimal or absent urticaria. All patients require two epinephrine autoinjectors and education on use. Venom immunotherapy is highly effective for Hymenoptera sting anaphylaxis.
Osteoporosis and Fractures
Present in 30-50% of mastocytosis patients, osteoporosis results from mast cell mediators affecting bone remodelling. [18] Vertebral compression fractures are the most common manifestation. All patients should undergo DEXA scanning at diagnosis and periodically. Treatment includes calcium, vitamin D, weight-bearing exercise, and bisphosphonates. Vertebroplasty may be required for symptomatic fractures.
Progression and Transformation
Indolent SM has a low risk of progression to advanced disease (5-10% over 20 years). [8] Factors associated with progression include high baseline tryptase (> 200 ng/mL), lack of skin involvement, presence of B-findings, and acquisition of additional mutations (TET2, SRSF2, ASXL1). Transformation to mast cell leukaemia is rare (1-2%) but carries an extremely poor prognosis.
10. Prognosis
Prognosis in systemic mastocytosis is highly dependent on disease variant.
Indolent SM (ISM)
- Life expectancy: Normal to near-normal; most patients do not experience shortened survival [8]
- Quality of life: Variable; many patients have good quality of life with appropriate symptom management; others are significantly impaired by mediator symptoms
- Progression risk: 5-10% progress to advanced SM over 20 years [8]
- Anaphylaxis risk: Significant cause of morbidity and potential mortality; lifelong risk requires vigilance
Smouldering SM (SSM)
- Life expectancy: Slightly reduced compared to ISM; higher risk of progression
- Progression: 10-20% progress to advanced SM over 10-20 years
- Monitoring: Requires closer follow-up than ISM
Advanced SM (ASM/SM-AHN/MCL)
- ASM untreated: Median survival 3-5 years [8]
- ASM with KIT inhibitors: Median survival extended to 7-10 years with avapritinib/midostaurin [6,20]
- SM-AHN: Prognosis determined by both SM and the associated haematological neoplasm; median survival 2-5 years depending on AHN
- MCL: Extremely poor; median survival less than 6 months even with treatment [8]; HSCT may offer cure in selected cases
Prognostic Factors
Favourable:
- ISM variant
- Presence of urticaria pigmentosa
- Younger age at diagnosis
- Absence of C-findings
- Serum tryptase less than 200 ng/mL
- KIT D816V mutation alone (no additional mutations)
- Good response to KIT inhibitor therapy
Unfavourable:
- Advanced SM variant (ASM, MCL)
- Absence of skin involvement
- C-findings (organ damage)
- Serum tryptase > 200 ng/mL
- Additional mutations: TET2, SRSF2, ASXL1, RUNX1 [16]
- Thrombocytopenia (less than 100 × 10⁹/L)
- Age > 60 years
11. Key Guidelines and Evidence
Major Society Guidelines
European Competence Network on Mastocytosis (ECNM):
- Consensus criteria for diagnosis and classification (aligned with WHO 2016) [1]
- Management recommendations for different SM variants
- Guidance on anaphylaxis prevention and management [10]
American Academy of Allergy, Asthma & Immunology (AAAAI):
- Guidelines on anaphylaxis management in mastocytosis patients
- Recommendations on venom immunotherapy
National Comprehensive Cancer Network (NCCN):
- Guidelines for systemic mastocytosis management (updated annually)
- Treatment algorithms for ISM and advanced SM
Landmark Trials
Midostaurin in Advanced SM:
- Gotlib et al., N Engl J Med 2016: Phase II trial demonstrating 60% overall response rate in advanced SM; FDA approval based on this study [5]
Avapritinib in Advanced SM:
- DeAngelo et al., Blood 2021 (EXPLORER trial): Phase I study showing 75% overall response rate [6]
- Reiter et al., Lancet Haematol 2022 (PATHFINDER trial): Avapritinib superior to midostaurin in head-to-head comparison [20]
Examination Focus
Common Exam Questions
-
"A 55-year-old man presents with recurrent episodes of flushing, diarrhoea, and syncope. On examination, he has multiple brown macules on his trunk that become raised when rubbed. How would you investigate?"
-
"What are the WHO diagnostic criteria for systemic mastocytosis?"
-
"Describe the management of a patient with indolent systemic mastocytosis who experiences frequent episodes of flushing and abdominal pain."
-
"What is the significance of the KIT D816V mutation in systemic mastocytosis, and how does it influence treatment?"
-
"A patient with known systemic mastocytosis requires general anaesthesia for elective surgery. What precautions would you take?"
Viva Points
Opening Statement: "Systemic mastocytosis is a clonal haematological disorder characterised by pathological accumulation of neoplastic mast cells in extracutaneous tissues, classified as a myeloproliferative neoplasm by the WHO. It is caused by activating mutations in the KIT proto-oncogene, most commonly D816V in approximately 90% of cases, leading to constitutive mast cell proliferation and mediator release."
Key Facts to Mention:
- Epidemiology: Rare disease; incidence 0.5-1 per 100,000; adults predominantly affected (median age 50-60)
- Pathognomonic triad: Elevated serum tryptase > 20 ng/mL + bone marrow mast cell aggregates + KIT D816V mutation
- Clinical presentation: Two mechanisms - mediator release (flushing, GI symptoms, anaphylaxis) and organ infiltration (hepatosplenomegaly, cytopenias)
- Darier sign: Wheal and flare upon rubbing skin lesions; pathognomonic for cutaneous mastocytosis
- WHO criteria: 1 major + 1 minor OR 3 minor criteria for diagnosis
- Major: Multifocal dense mast cell aggregates (≥15 per aggregate)
- Minor: (1) > 25% spindle/atypical morphology, (2) KIT D816V mutation, (3) aberrant CD25/CD2 expression, (4) tryptase > 20 ng/mL
- Classification: ISM (70-80%; excellent prognosis), SSM (5-10%), SM-AHN (20-30%), ASM (5-10%; median survival 3-5 years), MCL (less than 1%; median survival less than 6 months)
- Treatment ISM: Symptom control with H1+H2 antihistamines, cromolyn for GI symptoms, epinephrine autoinjector for all patients
- Treatment advanced SM: KIT inhibitors - avapritinib (preferred) or midostaurin for D816V-positive disease; response rates 60-75%
- Anaphylaxis risk: 20-50% lifetime risk; triggers include Hymenoptera stings, medications (NSAIDs, opioids, anaesthetics), radiocontrast
- D816V mutation significance: Present in 85-95% of SM; confers imatinib resistance but sensitivity to midostaurin/avapritinib; minor criterion for diagnosis
Differential Diagnosis to Mention:
- Carcinoid syndrome (5-HIAA elevated, hepatic metastases, normal tryptase)
- Phaeochromocytoma (episodic hypertension, elevated metanephrines)
- Hereditary alpha-tryptasemia (familial, tryptase less than 20 ng/mL, normal bone marrow)
- Chronic urticaria (transient wheals, no pigmentation, normal tryptase)
Management Algorithm: "I would stratify management by disease variant. For indolent SM, the focus is symptom control with H1 and H2 antihistamines, mast cell stabilizers like cromolyn sodium for GI symptoms, trigger avoidance, and all patients should carry two epinephrine autoinjectors due to anaphylaxis risk. For advanced SM with organ damage, I would initiate cytoreductive therapy with a KIT inhibitor - avapritinib is now preferred over midostaurin based on the PATHFINDER trial showing superior response rates in D816V-positive disease. Additionally, osteoporosis screening with DEXA and bisphosphonate therapy if indicated, and management of specific complications."
Red Flags/Complications:
- Anaphylaxis - severe, atypical presentation (minimal urticaria, profound hypotension)
- Progression to advanced SM - increasing tryptase, new cytopenias, organomegaly
- Transformation to mast cell leukaemia - rapidly progressive, multi-organ failure
Evidence to Quote:
- "The KIT D816V mutation is present in approximately 90% of systemic mastocytosis cases and is a WHO minor diagnostic criterion. [2]"
- "Midostaurin, approved by the FDA in 2017, demonstrated a 60% overall response rate in advanced SM in the pivotal Phase II trial by Gotlib et al. [5]"
- "Avapritinib showed superior efficacy to midostaurin in the PATHFINDER trial with overall response rates of 75%. [20]"
- "Anaphylaxis occurs in 20-50% of mastocytosis patients during their lifetime, with Hymenoptera stings being the most common trigger. [10]"
Common Mistakes to Avoid:
- ❌ Diagnosing SM based on elevated tryptase alone without bone marrow confirmation
- ❌ Using imatinib for D816V-positive SM (it is resistant; use midostaurin or avapritinib)
- ❌ Failing to prescribe epinephrine autoinjectors for all SM patients
- ❌ Not screening for osteoporosis (present in 30-50%)
- ❌ Forgetting perioperative premedication and anaesthetic agent selection
Model Answers
Q: A 45-year-old woman presents with a 5-year history of recurrent episodes of flushing, abdominal cramping, and diarrhoea. She has multiple brown macules on her trunk that she has noticed for 10 years. Describe your approach to investigating this patient.
A: "This presentation is highly suggestive of systemic mastocytosis. The combination of mediator-release symptoms (flushing, GI symptoms) and long-standing cutaneous lesions that sound like urticaria pigmentosa points towards this diagnosis. I would first examine the skin lesions and test for Darier sign by gently rubbing a lesion and observing for wheal and flare within 2-5 minutes, which would be pathognomonic.
My initial investigations would include serum tryptase, which if persistently elevated above 20 ng/mL is a major criterion for SM and would warrant further investigation. I would also perform a complete blood count to assess for cytopenias, liver and renal function tests, and consider differential diagnoses such as carcinoid syndrome by checking urinary 5-HIAA and chromogranin A.
If the tryptase is elevated, I would proceed to bone marrow biopsy, which is the gold standard for diagnosis. I would request histology looking for multifocal mast cell aggregates of ≥15 cells, immunohistochemistry for aberrant CD25 and CD2 expression on mast cells, and KIT mutation analysis to detect the D816V mutation present in 90% of cases. Additional investigations would include DEXA scan for osteoporosis screening, and imaging of the abdomen to assess for hepatosplenomegaly.
The WHO diagnostic criteria require either 1 major plus 1 minor criterion, or 3 minor criteria. Based on the findings, I would classify the disease variant and institute appropriate management, which for indolent SM would focus on symptom control with H1 and H2 antihistamines, cromolyn for GI symptoms, and provision of epinephrine autoinjectors given the significant anaphylaxis risk in mastocytosis patients."
Q: What is the significance of the KIT D816V mutation in systemic mastocytosis?
A: "The KIT D816V mutation is an activating mutation in the KIT proto-oncogene and is present in approximately 85-95% of adult systemic mastocytosis cases. It has several important clinical implications:
First, it is a WHO minor diagnostic criterion for SM. Its detection supports the diagnosis when combined with other criteria.
Second, it has pathophysiological significance as the D816V substitution in the kinase domain results in constitutive, ligand-independent activation of the KIT receptor tyrosine kinase. This drives mast cell proliferation, survival, and resistance to apoptosis through activation of downstream signalling pathways including RAS/RAF/MEK/ERK, PI3K/AKT, and JAK/STAT.
Third, and critically for treatment, the D816V mutation confers resistance to imatinib, which only binds to the inactive conformation of KIT. However, it confers sensitivity to Type II KIT inhibitors such as midostaurin and highly selective inhibitors like avapritinib, both of which are now FDA-approved for advanced SM. Therefore, KIT mutation testing is essential before initiating targeted therapy.
Fourth, it has prognostic implications. Patients with D816V as the sole mutation generally have a better prognosis than those who acquire additional mutations in genes such as TET2, SRSF2, or ASXL1, which are associated with disease progression and adverse outcomes.
Finally, the mutation can be detected in bone marrow, peripheral blood, or skin biopsy specimens, although bone marrow is the most sensitive source."
References
-
Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420-1427. doi:10.1182/blood-2016-09-731893
-
Kristensen T, Vestergaard H, Bindslev-Jensen C, et al. Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493-498. doi:10.1002/ajh.23672
-
Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis. J Allergy Clin Immunol. 2016;137(1):35-45. doi:10.1016/j.jaci.2015.08.034
-
Matito A, Azaña JM, Torrelo A, et al. Cutaneous mastocytosis in adults and children: New classification and prognostic factors. Immunol Allergy Clin North Am. 2018;38(3):351-363. doi:10.1016/j.iac.2018.04.001
-
Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530-2541. doi:10.1056/NEJMoa1513098
-
DeAngelo DJ, Quiery AT, Radia D, et al. Clinical activity in a phase 1 study of avapritinib in patients with advanced systemic mastocytosis. Blood. 2021;138(Suppl 1):2246. doi:10.1182/blood-2021-147955
-
Cohen SS, Skovbo S, Vestergaard H, et al. Epidemiology of systemic mastocytosis in Denmark. Br J Haematol. 2014;166(4):521-528. doi:10.1111/bjh.12916
-
Lim KH, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood. 2009;113(23):5727-5736. doi:10.1182/blood-2009-02-205237
-
Schwartz LB. Clinical utility of tryptase levels in systemic mastocytosis and associated hematologic disorders. Leuk Res. 2001;25(7):553-562. doi:10.1016/s0145-2126(01)00050-1
-
Bonadonna P, Pagani M, Aberer W, et al. Drug hypersensitivity in clonal mast cell disorders: ENDA/EAACI position paper. Allergy. 2015;70(7):755-763. doi:10.1111/all.12617
-
Horny HP, Sotlar K, Valent P. Mastocytosis: State of the art. Pathobiology. 2007;74(2):121-132. doi:10.1159/000101711
-
Pardanani A. Systemic mastocytosis in adults: 2017 update on diagnosis, risk stratification and management. Am J Hematol. 2016;91(11):1146-1159. doi:10.1002/ajh.24553
-
Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30(4):373-378. doi:10.1016/j.leukres.2005.08.015
-
Jawhar M, Schwaab J, Schnittger S, et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V+ advanced systemic mastocytosis. Leukemia. 2016;30(1):136-143. doi:10.1038/leu.2015.284
-
Arock M, Sotlar K, Akin C, et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia. 2015;29(6):1223-1232. doi:10.1038/leu.2015.24
-
Jawhar M, Schwaab J, Hausmann D, et al. Splenomegaly, elevated alkaline phosphatase and mutations in the SRSF2/ASXL1/RUNX1 gene panel are strong adverse prognostic markers in patients with systemic mastocytosis. Leukemia. 2016;30(12):2342-2350. doi:10.1038/leu.2016.190
-
Sokol H, Georgin-Lavialle S, Canioni D, et al. Gastrointestinal manifestations in mastocytosis: A study of 83 patients. J Allergy Clin Immunol. 2013;132(4):866-873. doi:10.1016/j.jaci.2013.05.026
-
Barete S, Assous N, de Gennes C, et al. Systemic mastocytosis and bone involvement in a cohort of 75 patients. Ann Rheum Dis. 2010;69(10):1838-1841. doi:10.1136/ard.2009.124511
-
Kibsgaard L, Skjold T, Deleuran M, Vestergaard C. Omalizumab induced remission of idiopathic anaphylaxis in a patient with indolent systemic mastocytosis. Acta Derm Venereol. 2014;94(3):363-364. doi:10.2340/00015555-1698
-
Reiter A, Gotlib J, Alvarez-Twose I, et al. Efficacy of avapritinib versus best available therapy in the treatment of advanced systemic mastocytosis. Lancet Haematol. 2022;9(5):e328-e339. doi:10.1016/S2352-3026(22)00069-7
Last Updated: 2026-01-05
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Differentials
Competing diagnoses and look-alikes to compare.
- Anaphylaxis
- Urticaria
- Carcinoid Syndrome
- Phaeochromocytoma