Mixed Connective Tissue Disease
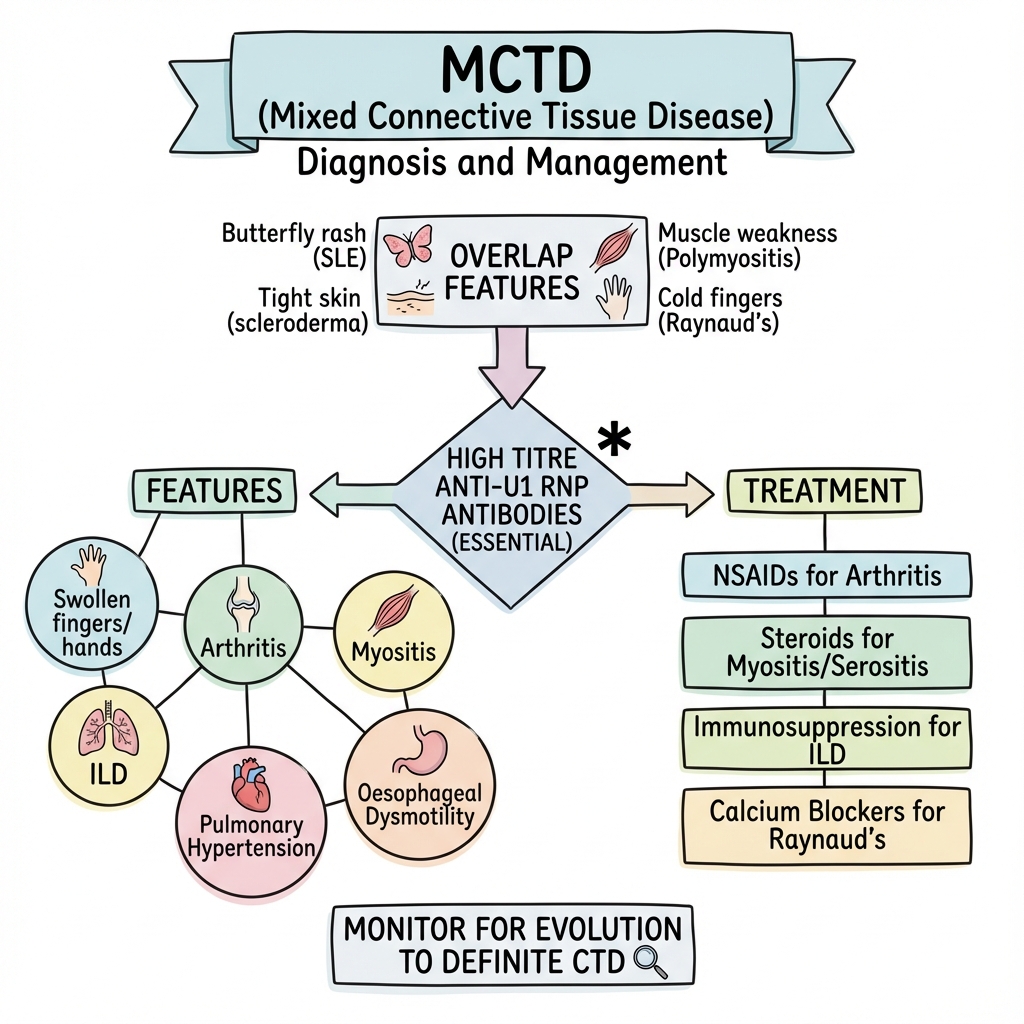
The defining serological hallmark is the presence of high-titre anti-U1 RNP antibodies, which are essential for diagnosis. Clinical manifestations typically evolve over time, with early disease characterized by...
What matters first
The defining serological hallmark is the presence of high-titre anti-U1 RNP antibodies, which are essential for diagnosis. Clinical manifestations typically evolve over time, with early disease characterized by...
Pulmonary arterial hypertension
6 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Pulmonary arterial hypertension
- Rapidly progressive ILD
- Severe myositis
- Evolution to severe SLE or scleroderma
Linked comparisons
Differentials and adjacent topics worth opening next.
- Undifferentiated Connective Tissue Disease
- Overlap Syndromes
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Mixed Connective Tissue Disease
1. Overview
Mixed connective tissue disease (MCTD) is a distinct overlap syndrome characterized by clinical features of systemic lupus erythematosus (SLE), systemic sclerosis (SSc), and polymyositis (PM), occurring in association with high-titre antibodies to U1 small nuclear ribonucleoprotein (anti-U1 RNP). [1] First described by Gordon Sharp in 1972, MCTD was initially thought to represent a "benign" overlap syndrome with relative sparing of renal disease. [2] However, subsequent long-term studies have demonstrated significant morbidity and mortality, particularly related to pulmonary arterial hypertension (PAH) and interstitial lung disease (ILD). [3]
The defining serological hallmark is the presence of high-titre anti-U1 RNP antibodies, which are essential for diagnosis. Clinical manifestations typically evolve over time, with early disease characterized by Raynaud's phenomenon, puffy hands, and arthralgias, progressing to more specific organ involvement including inflammatory myopathy, pulmonary complications, and oesophageal dysmotility. [4] The disease predominantly affects women (female to male ratio 9:1) with peak onset between the third and fifth decades of life. [5]
MCTD occupies a controversial position in rheumatology nosology. While some authorities consider it a distinct entity, others view it as part of the spectrum of undifferentiated connective tissue disease (UCTD) or simply an overlap syndrome. [6] The key clinical importance lies in recognizing that approximately 30% of patients initially diagnosed with MCTD will eventually evolve into a definite connective tissue disease (most commonly SSc or SLE), and that early identification and monitoring for PAH is critical for reducing mortality. [7]
2. Epidemiology
Incidence and Prevalence
The reported incidence of MCTD varies widely across studies, ranging from 2.0 to 10 per 100,000 population, with prevalence estimates between 3.8 and 10 per 100,000. [8] This variation reflects both true geographic differences and inconsistent application of diagnostic criteria. Population-based studies from Scandinavia suggest a prevalence of approximately 3.8 per 100,000, while Japanese cohorts report higher figures. [9]
| Statistic | Value | Source |
|---|---|---|
| Prevalence | 3.8-10 per 100,000 | [8] |
| Annual incidence | 2.0 per 100,000 | [9] |
| Female to male ratio | 9:1 | [5] |
| Mean age at diagnosis | 37 years (range 4-80) | [10] |
| 10-year survival | 80-90% | [11] |
| Evolution to definite CTD | 30% | [7] |
Demographics and Risk Factors
Age and Sex: MCTD predominantly affects women of childbearing age, with peak incidence between 30-50 years, although cases have been described from childhood to late adulthood. [5] The overwhelming female predominance (approximately 90% of cases) suggests hormonal influences, similar to other systemic autoimmune rheumatic diseases. [10]
Ethnicity: Unlike SLE, which shows marked ethnic variation with higher prevalence in African-American and Hispanic populations, MCTD appears to have a more uniform distribution across ethnic groups. However, limited epidemiological data from diverse populations makes definitive conclusions difficult. [12]
Genetic Susceptibility: HLA associations in MCTD are less robust than in other connective tissue diseases. Some studies have reported associations with HLA-DR4, HLA-DR2, and HLA-DRw53, but these findings have not been consistently replicated. [13] Family clustering is rare, distinguishing MCTD from diseases with stronger genetic components such as SLE.
3. Aetiology and Pathophysiology
Molecular Basis of Anti-U1 RNP Autoimmunity
The U1 small nuclear ribonucleoprotein (U1 snRNP) is a component of the spliceosome complex responsible for pre-mRNA splicing. The U1 snRNP particle consists of U1 RNA and 10 associated proteins, including U1-70 kDa, U1-A, and U1-C proteins. [14] In MCTD, autoantibodies target epitopes on these proteins, most commonly the U1-70 kDa protein.
The pathogenic mechanisms underlying anti-U1 RNP-mediated disease remain incompletely understood. Several hypotheses have been proposed:
-
Molecular Mimicry: Environmental triggers (potentially viral infections) may present epitopes that cross-react with U1 snRNP components, initiating an autoimmune response in genetically susceptible individuals. [15]
-
Apoptotic Dysregulation: Enhanced apoptosis leading to surface blebs enriched in U1 snRNP may provide autoantigens in an immunogenic context, breaking self-tolerance. [16]
-
Interferon Signature: Like SLE, MCTD patients demonstrate an interferon-alpha gene signature, suggesting innate immune dysregulation contributes to disease pathogenesis. [17]
-
Immune Complex Deposition: Anti-U1 RNP antibodies can form immune complexes that deposit in target tissues, triggering complement activation and inflammatory responses.
Exam Detail: Detailed Immunopathogenesis:
The U1 snRNP complex contains multiple antigenic targets:
- U1-70K (70 kDa protein): Most common target, present in > 95% of MCTD patients
- U1-A (34 kDa protein): Associated with more severe disease manifestations
- U1-C (22 kDa protein): Correlates with specific clinical phenotypes
Anti-U1 RNP antibodies in MCTD are predominantly IgG class, with all four subclasses represented. The antibody titre and epitope specificity may correlate with disease activity and organ involvement, though this remains controversial.
Cytokine Profile: MCTD demonstrates a mixed Th1/Th2 cytokine pattern:
- Elevated IL-6, IL-10, and TNF-α during active disease
- Interferon-regulated genes are upregulated
- IL-17 may contribute to pulmonary vascular remodelling in PAH
Vascular Pathology: The microvascular injury characteristic of MCTD involves:
- Endothelial cell activation and damage
- Smooth muscle cell proliferation
- Adventitial fibroblast activation
- Perivascular inflammatory infiltrates
- Progressive luminal narrowing
This vasculopathy is central to Raynaud's phenomenon, digital ischaemia, and pulmonary arterial hypertension development.
Organ-Specific Pathology
Pulmonary: PAH in MCTD results from a proliferative vasculopathy affecting small pulmonary arterioles, with intimal hyperplasia, medial hypertrophy, adventitial fibrosis, and in situ thrombosis. This process is indistinguishable from idiopathic PAH histologically. ILD typically shows a nonspecific interstitial pneumonia (NSIP) pattern, less commonly usual interstitial pneumonia (UIP). [18]
Musculoskeletal: Inflammatory myopathy demonstrates lymphocytic infiltration of muscle fibres, perivascular inflammation, and variable degrees of muscle fibre necrosis and regeneration. Synovitis is typically non-erosive, resembling SLE more than rheumatoid arthritis. [19]
Renal: Unlike SLE, severe proliferative glomerulonephritis is rare in MCTD. When present, renal involvement typically manifests as membranous nephropathy or mild mesangial disease. [20]
4. Clinical Presentation
Evolution and Natural History
MCTD typically evolves through several phases:
Phase 1 - Early/Undifferentiated Phase (months to years):
- Raynaud's phenomenon (often the initial manifestation)
- Puffy hands/fingers ("sausage digits")
- Arthralgias and mild arthritis
- General constitutional symptoms (fatigue, low-grade fever)
- Positive anti-U1 RNP but incomplete criteria for definite MCTD
Phase 2 - Overlap Phase (years):
- Progressive accumulation of features from multiple CTDs
- Development of myositis, serositis, or ILD
- Oesophageal involvement
- More definite overlap syndrome emerges
Phase 3 - Late/Complication Phase (5-15 years):
- PAH development (the major cause of morbidity/mortality)
- Progressive ILD
- Potential evolution to definite SSc or SLE (30% of cases)
- End-organ complications
Cardinal Clinical Features
Raynaud's Phenomenon (95%)
Raynaud's is almost universal in MCTD, often preceding other manifestations by years. It tends to be more severe than in primary Raynaud's, with pronounced colour changes (triphasic response) and higher risk of digital ulceration and gangrene. [21] Nailfold capillaroscopy reveals abnormalities intermediate between SLE (preserved capillary density with some architectural disruption) and SSc (severe capillary loss and giant loops).
Swollen Hands (90%)
Diffuse, non-pitting oedema of the hands and fingers produces characteristic "sausage fingers" or "puffy hands." This feature, often symmetric and painless, reflects a combination of inflammation and early vascular changes. It may be the most specific early clinical clue to MCTD when combined with high-titre anti-U1 RNP antibodies. [22]
Arthritis and Arthralgia (80%)
Joint involvement ranges from migratory arthralgias to frank symmetric polyarthritis affecting small joints of hands, wrists, and knees. Unlike rheumatoid arthritis, erosive disease is uncommon (reported in less than 10% of cases). Radiographic findings, when present, typically show juxta-articular osteopenia and joint space narrowing without marginal erosions. Deforming arthropathy (Jaccoud's arthropathy) may develop. [23]
Myositis (50-70%)
Muscle involvement manifests with:
- Proximal muscle weakness (difficulty climbing stairs, rising from chairs)
- Myalgia (muscle pain and tenderness)
- Elevated muscle enzymes (CK, aldolase, LDH, AST, ALT)
- Abnormal EMG (myopathic pattern with fibrillations)
- MRI changes (muscle oedema on STIR sequences)
- Muscle biopsy (inflammatory infiltrates, fibre necrosis)
The myositis in MCTD is clinically and histologically similar to polymyositis but may be more responsive to corticosteroid therapy. [19]
Organ System Involvement
| Organ System | Manifestation | Frequency | Clinical Significance |
|---|---|---|---|
| Pulmonary | PAH | 10-25% | Leading cause of mortality |
| ILD (NSIP pattern) | 50-65% | Progressive in 10-20% | |
| Pleuritis | 30-50% | Usually mild, responsive to therapy | |
| Cardiac | Pericarditis | 30% | Typically asymptomatic |
| Myocarditis | 10% | Rare but serious | |
| Gastrointestinal | Oesophageal dysmotility | 50-80% | Similar to SSc pattern |
| Gastric antral vascular ectasia | 5% | Rare but causes bleeding | |
| Renal | Glomerulonephritis | 10% | Usually mild, not like SLE |
| Membranous nephropathy | 5-10% | Proteinuria predominant | |
| Neurological | Trigeminal neuropathy | 10-25% | Characteristic feature |
| Aseptic meningitis | less than 5% | Resembles SLE | |
| Cognitive dysfunction | Variable | Underrecognized | |
| Haematological | Anaemia | 50% | Usually ACD pattern |
| Leucopenia | 20-30% | Mild, rarely clinically significant | |
| Thrombocytopenia | 20% | Mild, rarely less than 50 × 10⁹/L |
Red Flag Features
Certain clinical presentations demand urgent assessment:
- Progressive dyspnoea: May herald PAH or rapidly progressive ILD - requires urgent TTE and PFTs
- Severe proximal weakness: Suggests aggressive myositis requiring high-dose immunosuppression
- Acute chest pain: Consider pericarditis, myocarditis, or pulmonary embolism
- Digital gangrene: Severe vascular compromise requiring vasodilator therapy and possible surgical intervention
- Acute neurological symptoms: Excludes secondary causes (infection, malignancy) and may require aggressive immunosuppression
5. Differential Diagnosis
The diagnosis of MCTD requires careful exclusion of other conditions in the spectrum of systemic autoimmune rheumatic diseases:
1. Undifferentiated Connective Tissue Disease (UCTD)
Key Distinguishing Features:
- UCTD patients have incomplete criteria for any specific CTD
- Anti-U1 RNP antibodies may be present but typically at lower titres
- Organ involvement is less severe
- Many UCTD patients remain stable or remit; MCTD typically progresses
- UCTD does not meet formal diagnostic criteria for MCTD (see Diagnostic Criteria section)
2. Systemic Lupus Erythematosus
Key Distinguishing Features:
- SLE typically has anti-dsDNA and anti-Sm antibodies; anti-U1 RNP alone is insufficient for SLE diagnosis
- Renal disease (proliferative GN) and CNS lupus are common in SLE, rare in MCTD
- Severe haematological abnormalities more common in SLE
- Photosensitivity and malar rash more characteristic of SLE
- However, 30% of MCTD may evolve into SLE
3. Systemic Sclerosis
Key Distinguishing Features:
- SSc has anti-Scl-70 (diffuse cutaneous) or anti-centromere (limited cutaneous) antibodies
- Skin thickening extending proximal to MCPs in diffuse SSc
- Severe vascular disease with extensive digital pitting scars
- More severe and progressive ILD
- Renal crisis characteristic of SSc, not MCTD
- However, MCTD can evolve into SSc
4. Polymyositis/Dermatomyositis
Key Distinguishing Features:
- PM/DM have myositis-specific antibodies (anti-Jo-1, anti-Mi-2, anti-TIF1-γ, etc.)
- Heliotrope rash and Gottron's papules in dermatomyositis
- Higher malignancy risk in DM (especially anti-TIF1-γ positive)
- Less multisystem involvement
5. Rheumatoid Arthritis with Extra-articular Features
Key Distinguishing Features:
- Positive RF and anti-CCP antibodies
- Erosive arthritis on imaging
- Less prominent Raynaud's phenomenon
- Different pattern of organ involvement
| Differential | Anti-U1 RNP | Distinguishing Antibodies | Key Clinical Differences |
|---|---|---|---|
| MCTD | High titre (essential) | None specific | Overlap features, Raynaud's, puffy hands |
| SLE | Low-moderate titre (30%) | Anti-dsDNA, anti-Sm | Renal, CNS disease, photosensitivity |
| SSc | Negative/low titre | Anti-Scl-70, anti-centromere | Skin thickening, scleroderma pattern |
| PM/DM | Negative | Anti-Jo-1, anti-SRP, anti-Mi-2 | Isolated myositis, heliotrope rash |
| UCTD | Variable titre | None diagnostic | Incomplete criteria, milder disease |
6. Investigations
Serological Testing
Essential Investigations
Anti-U1 RNP Antibodies:
- Test: ELISA or immunoblot
- Threshold: High titre required for MCTD diagnosis (typically > 1:1600 or > 100 units)
- Sensitivity: 95-100% (defines the disease)
- Specificity: Moderate (present in 30% of SLE, 10% of SSc)
- Clinical correlation: Titre may correlate with disease activity in some patients
Antinuclear Antibodies (ANA):
- Pattern: Speckled nuclear pattern on immunofluorescence
- Titre: Universally positive, typically ≥1:320
- Significance: Screening test; anti-U1 RNP confirmation required
Supportive Serology
| Test | Finding in MCTD | Interpretation |
|---|---|---|
| Anti-dsDNA | Usually negative | If positive, suggests SLE overlap |
| Anti-Sm | Negative | Helps distinguish from SLE |
| Anti-Scl-70 | Negative | Helps distinguish from diffuse SSc |
| Anti-centromere | Negative | Helps distinguish from limited SSc |
| Rheumatoid factor | Positive (50-70%) | Non-specific, does not indicate RA |
| Anti-CCP | Negative | Helps exclude RA |
| Complement (C3, C4) | Normal or mildly reduced | Severe reduction suggests SLE |
| ESR/CRP | Elevated during active disease | Correlates with inflammation |
Organ-Specific Investigations
Pulmonary Assessment (Critical for PAH Screening)
Baseline and Annual Screening:
-
Pulmonary Function Tests:
- FVC and DLCO (reduced in ILD and PAH)
- DLCO less than 70% predicted is red flag for PAH
- Isolated DLCO reduction without restrictive pattern suggests vascular disease
-
Transthoracic Echocardiography:
- Estimated right ventricular systolic pressure (RVSP)
- RVSP > 40 mmHg warrants further investigation
- Right heart function and size assessment
- Pericardial effusion detection
-
High-Resolution CT Chest (if ILD suspected):
- NSIP pattern most common (ground-glass opacification, reticular changes)
- UIP pattern less common
- Extent of involvement guides prognosis
-
Right Heart Catheterization (gold standard for PAH diagnosis):
- Indications: RVSP > 40 mmHg on echo, unexplained dyspnoea, DLCO less than 60%
- Diagnostic criteria: mPAP ≥25 mmHg, PCWP ≤15 mmHg, PVR > 3 Wood units (updated criteria now use mPAP ≥20 mmHg)
Musculoskeletal Assessment
For Suspected Myositis:
- Creatine Kinase: Elevated (often 5-50× normal during active myositis)
- Aldolase, LDH, AST, ALT: Support diagnosis when CK equivocal
- Electromyography: Myopathic changes (short-duration, low-amplitude polyphasic potentials; fibrillation potentials)
- MRI Muscle: T2/STIR hyperintensity indicates muscle oedema/inflammation; guides biopsy site
- Muscle Biopsy: Endomysial/perimysial inflammatory infiltrates, variable fibre necrosis
For Arthritis:
- Plain Radiographs: Non-erosive arthritis, juxta-articular osteopenia
- Ultrasound/MRI: Synovitis and tenosynovitis detection
Gastrointestinal Assessment
- Barium Swallow/Oesophageal Manometry: Reduced lower oesophageal sphincter pressure, dysmotility similar to SSc
- Upper GI Endoscopy: If GORD symptoms, exclude Barrett's oesophagus
Renal Assessment
- Urinalysis: Proteinuria, haematuria (if present, suggests GN)
- Serum Creatinine and eGFR: Baseline renal function
- Urine Protein:Creatinine Ratio: Quantify proteinuria
- Renal Biopsy: If significant proteinuria or active urinary sediment
Haematological Assessment
- Full Blood Count: Anaemia, leucopenia, thrombocytopenia (usually mild)
- Blood Film: Exclude haemolysis (unlike SLE, AIHA rare in MCTD)
Nailfold Capillaroscopy
- Findings intermediate between SLE and SSc
- Some capillary loss, architectural disruption, occasional giant loops
- Useful for risk stratification and monitoring
7. Diagnostic Criteria
No universally accepted diagnostic criteria exist for MCTD, contributing to ongoing controversy about its status as a distinct entity. Three classification systems are most commonly cited:
Alarcon-Segovia Criteria (1987)
Requires:
- Positive anti-U1 RNP antibodies (high titre)
Plus 3 of the following clinical features:
- Oedema of the hands
- Synovitis
- Myositis
- Raynaud's phenomenon
- Acrosclerosis (SSc-like skin changes)
Exclusions: Scleroderma proximal to MCPs, malar rash, discoid lupus, photosensitivity, oral ulcers, pleuritis, pericarditis, casts, CNS involvement, haemolytic anaemia, leucopenia, thrombocytopenia
This is the most restrictive criteria, emphasizing a "pure" phenotype. [4]
Kasukawa Criteria (1987)
Requires:
- Positive anti-U1 RNP antibodies
Plus at least one of:
- Raynaud's phenomenon
- Swollen fingers/hands
Plus two or more of:
- SLE-like features (polyarthritis, lymphadenopathy, facial erythema, pericarditis/pleuritis, leucopenia/thrombocytopenia)
- SSc-like features (sclerodactyly, pulmonary fibrosis, dysmotility/dilatation of oesophagus)
- PM-like features (muscle weakness, elevated CK, myogenic pattern on EMG)
This is more inclusive and commonly used in Japanese studies. [24]
Kahn Criteria (1989)
Major Criteria (all required):
- Positive anti-U1 RNP antibodies
- Myositis
- Pulmonary involvement (DLCO less than 70% or PAH or ILD)
- Raynaud's phenomenon or oesophageal dysmotility
- Swollen hands or sclerodactyly
This emphasizes severe organ involvement and is less commonly used.
Practical Diagnostic Approach
In clinical practice, most rheumatologists diagnose MCTD when:
- High-titre anti-U1 RNP antibodies are present (essential)
- Overlap features from at least 2-3 of SLE, SSc, PM are evident
- Raynaud's phenomenon is present (almost universal)
- Alternative diagnoses (definite SLE, SSc, PM) are excluded
It is important to recognize MCTD as a provisional diagnosis that may evolve over time.
8. Management
MCTD management is symptom-directed and organ-based, as no disease-modifying therapy specific to MCTD exists. The approach mirrors management strategies for SLE, SSc, and PM, tailored to the individual patient's manifestations.
General Principles
- Early identification and monitoring of PAH: Leading cause of mortality
- Aggressive treatment of myositis and ILD: Prevent irreversible damage
- Cardiovascular risk reduction: Chronic inflammation increases CV risk
- Patient education: Self-monitoring for disease evolution
- Multidisciplinary care: Rheumatology, pulmonology, cardiology collaboration
Manifestation-Specific Treatment
Raynaud's Phenomenon and Digital Ischaemia
Conservative Measures:
- Smoking cessation (essential)
- Cold avoidance, protective clothing
- Stress management
Pharmacological:
- First-line: Calcium channel blockers (nifedipine 30-60 mg/day extended-release, amlodipine 5-10 mg/day)
- Second-line: Phosphodiesterase-5 inhibitors (sildenafil 20 mg TDS, tadalafil 20 mg daily)
- Refractory: Prostanoids (iloprost IV infusions), endothelin receptor antagonists (bosentan)
Digital Ulcers/Gangrene:
- IV prostacyclin (iloprost 0.5-2 ng/kg/min for 3-5 days)
- Bosentan 62.5 mg BD → 125 mg BD
- Surgical debridement if gangrenous
Arthritis and Arthralgias
Mild:
- NSAIDs (naproxen 500 mg BD, ibuprofen 400-600 mg TDS)
- Low-dose prednisolone (5-10 mg/day)
Moderate-Severe:
- Hydroxychloroquine 200-400 mg/day (safe, anti-inflammatory)
- Methotrexate 15-25 mg/week (if hydroxychloroquine insufficient)
- Azathioprine 1-2 mg/kg/day (alternative DMARD)
Refractory:
- Biological therapies (anti-TNF, rituximab) - limited evidence in MCTD
Myositis
Acute/Severe:
- High-dose corticosteroids: Prednisolone 1 mg/kg/day (max 60-80 mg/day) for 4-6 weeks, then gradual taper over 12-18 months
- Steroid-sparing agents (initiated early):
- Methotrexate 15-25 mg/week (first-line)
- Azathioprine 2-3 mg/kg/day (alternative)
- Mycophenolate mofetil 2-3 g/day (increasingly used)
Refractory:
- IV immunoglobulin (2 g/kg over 2-5 days monthly)
- Rituximab (1 g IV on days 0 and 14, or 375 mg/m² weekly ×4)
- Calcineurin inhibitors (tacrolimus, ciclosporin)
Monitoring:
- CK levels (initially weekly, then monthly)
- Muscle strength assessment (MRC grading)
- Serial MRI if equivocal response
Interstitial Lung Disease
Mild, Stable:
- Observation with serial PFTs and HRCT
Moderate-Severe or Progressive:
- Corticosteroids: Prednisolone 0.5-1 mg/kg/day for 4-6 weeks, then taper
- Immunosuppression:
- Mycophenolate mofetil 1.5-3 g/day (preferred agent based on SSc-ILD data)
- Cyclophosphamide IV (500-750 mg/m² monthly for 6-12 months) - for rapidly progressive ILD
- Azathioprine 2 mg/kg/day (maintenance therapy)
Monitoring:
- PFTs every 3-6 months (FVC, DLCO)
- HRCT at baseline and if clinical deterioration
Pulmonary Arterial Hypertension
Approach parallels idiopathic PAH management:
General Measures:
- Oxygen therapy if hypoxic (SpO₂ less than 90%)
- Diuretics for fluid overload
- Anticoagulation (controversial in CTD-PAH; individualize based on risk/benefit)
- Immunosuppression (case series suggest benefit in some MCTD-PAH patients)
PAH-Specific Therapies (initiated by pulmonary hypertension specialist):
Mild PAH (WHO FC I-II):
- Phosphodiesterase-5 inhibitors (sildenafil 20 mg TDS, tadalafil 40 mg daily)
- Endothelin receptor antagonists (bosentan 125 mg BD, ambrisentan 5-10 mg daily, macitentan 10 mg daily)
Moderate-Severe PAH (WHO FC III-IV):
- Combination therapy (PDE5i + ERA)
- Prostacyclin analogues (epoprostenol IV, treprostinil SC/IV, iloprost inhaled)
- Riociguat (soluble guanylate cyclase stimulator)
Refractory:
- Lung transplantation assessment
Monitoring:
- 6-minute walk test, BNP/NT-proBNP, echo every 3-6 months
- Right heart catheterization to assess response to therapy
Oesophageal Dysmotility
- PPI therapy: Omeprazole 20-40 mg daily, lansoprazole 30 mg daily (prevent reflux complications)
- Prokinetics: Metoclopramide 10 mg TDS (limited efficacy)
- Lifestyle: Small frequent meals, avoid late-night eating, head elevation
Serositis (Pericarditis/Pleuritis)
- NSAIDs: High-dose with gastroprotection
- Corticosteroids: Prednisolone 20-40 mg/day if NSAIDs insufficient, taper over 4-8 weeks
- Colchicine: 0.5-1 mg/day (prevent recurrence)
Renal Involvement
- Mild proteinuria (less than 1 g/day): ACEI/ARB for renoprotection, monitoring
- Nephrotic syndrome or GN: Corticosteroids + immunosuppression (cyclophosphamide or mycophenolate), as per SLE nephritis protocols
Pregnancy in MCTD
Preconception Counselling:
- Optimize disease control (quiescent for ≥6 months preferred)
- Medication review (stop methotrexate, mycophenolate, cyclophosphamide; continue hydroxychloroquine, azathioprine if needed)
- Screen for PAH (contraindication to pregnancy if moderate-severe)
- Check anti-Ro/La antibodies (risk of neonatal lupus, congenital heart block)
Antenatal Management:
- Multidisciplinary care (rheumatology, obstetrics, cardiology)
- Serial echocardiograms if anti-Ro positive (monitor for fetal heart block)
- Low-dose aspirin (reduce pre-eclampsia risk)
- Monitor for disease flare (particularly post-partum)
Outcomes: Generally favorable in well-controlled disease without PAH; increased risk of preterm delivery, pre-eclampsia, and disease flare. [25]
Follow-Up and Monitoring
Baseline Assessment:
- Comprehensive serology, PFTs, HRCT chest, echo, ECG
- Baseline organ function (renal, hepatic, haematological)
- Baseline muscle strength if myositis suspected
Regular Monitoring:
- Clinical review: Every 3-6 months when stable, more frequently if active
- PFTs and echo: Annually (critical for early PAH detection)
- FBC, U&E, LFT: Every 3-6 months (medication monitoring)
- CK: If myositis, titrate monitoring to disease activity
Red Flags Requiring Urgent Assessment:
- Progressive dyspnoea (PAH, ILD progression)
- Significant proximal weakness (myositis flare)
- Unexplained decline in DLCO > 15%
- Acute neurological symptoms
9. Complications
| Complication | Frequency | Prevention | Management |
|---|---|---|---|
| Pulmonary arterial hypertension | 10-25% | Annual echo + PFT screening | PAH-specific therapies, immunosuppression |
| Progressive ILD | 10-20% of ILD patients | Early immunosuppression if active | Mycophenolate, cyclophosphamide |
| Digital gangrene | 5-10% | Vasodilator therapy, smoking cessation | IV iloprost, bosentan, surgical debridement |
| Myocarditis | 5-10% | Early recognition of cardiac symptoms | High-dose steroids, immunosuppression |
| Severe myositis | 5% | Early aggressive treatment | High-dose steroids, IVIG, rituximab |
| Cerebrovascular events | 3-5% | CV risk factor management | Standard stroke protocols + immunosuppression |
| Malignancy | Standard population risk | Age-appropriate screening | Not elevated unlike DM |
10. Prognosis
Survival and Outcomes
Overall prognosis in MCTD is intermediate between SLE and SSc, with 10-year survival rates of 80-90% in most series. [11] However, survival is highly dependent on organ involvement, particularly pulmonary disease.
Prognostic Factors:
Favourable:
- Limited organ involvement
- Absence of PAH and severe ILD
- Good response to initial therapy
- Younger age at onset
Unfavourable:
- PAH (single most important predictor of mortality)
- Progressive ILD
- Myocardial involvement
- Renal disease (though rare)
- Evolution to definite SSc
Causes of Death:
- PAH (leading cause, 30-50% of deaths)
- ILD with respiratory failure
- Cardiovascular disease (myocarditis, MI, CVA)
- Infection (particularly if heavily immunosuppressed)
- Malignancy (not elevated above baseline unlike DM)
Disease Evolution
Approximately 30% of patients initially diagnosed with MCTD will evolve into a definite connective tissue disease over 5-10 years of follow-up. [7]
Evolution patterns:
- To SSc (most common): Progressive skin thickening, worsening vascular disease, more extensive ILD
- To SLE: Development of anti-dsDNA antibodies, renal disease, CNS involvement
- To PM: Isolated myositis without overlap features
- Remain as MCTD: 70% maintain overlap syndrome phenotype
Monitoring for evolution:
- Serial serological testing
- Vigilance for new organ manifestations
- Adjust management strategy if definite disease emerges
11. Key Guidelines and Evidence
Major Society Guidelines:
- EULAR/ACR: No MCTD-specific guidelines; management extrapolated from SLE, SSc, myositis guidelines
- EULAR PAH Guidelines (2022): Recommend annual echo screening in all CTD patients for early PAH detection
- EULAR Myositis Guidelines (2016): Inform immunosuppressive strategies for MCTD-associated myositis
- ATS/ERS ILD Guidelines: Guide management of CTD-ILD
Landmark Studies:
- Sharp's original description (1972): Defined the syndrome [2]
- Alarcon-Segovia diagnostic criteria (1987): Most commonly cited classification [4]
- Long-term outcome studies: Established PAH as leading cause of mortality [3,11]
12. Exam-Focused Content
Common Exam Questions
-
"What are the diagnostic criteria for MCTD?"
- High-titre anti-U1 RNP antibodies (essential)
- Clinical features overlapping SLE, SSc, PM
- Most commonly use Alarcon-Segovia or Kasukawa criteria
- Raynaud's phenomenon almost universal
- Exclusion of definite alternative CTD diagnosis
-
"What is the leading cause of mortality in MCTD and how do you screen for it?"
- Pulmonary arterial hypertension (PAH)
- Screen with annual echocardiography (RVSP) and pulmonary function tests (DLCO)
- RVSP > 40 mmHg or unexplained DLCO reduction warrants right heart catheterization
- Early detection and PAH-specific therapy improves outcomes
-
"How would you manage a patient with MCTD presenting with progressive dyspnoea?"
- Systematic approach to differentiate PAH vs ILD vs cardiac vs other
- PFTs (DLCO isolated reduction suggests PAH, restrictive pattern suggests ILD)
- Echo (estimate RVSP, assess RV function)
- HRCT chest (ILD pattern)
- Right heart catheterization if PAH suspected
- Treatment: PAH-specific therapies (PDE5i, ERA, prostanoids) and immunosuppression for ILD (mycophenolate, cyclophosphamide)
-
"What is the natural history and prognosis of MCTD?"
- 10-year survival 80-90%
- 30% evolve to definite CTD (SSc or SLE most common)
- PAH is the leading cause of death
- Prognosis depends on organ involvement
- Better prognosis than SSc, similar or slightly worse than SLE
Viva Points
Viva Point: Opening Statement: "Mixed connective tissue disease is an overlap syndrome characterized by features of SLE, systemic sclerosis, and polymyositis, occurring in association with high-titre antibodies to U1 small nuclear ribonucleoprotein. It predominantly affects women with a 9:1 female to male ratio."
Key Facts to Mention:
- First described by Sharp in 1972
- Prevalence approximately 3.8-10 per 100,000
- Anti-U1 RNP antibodies are essential for diagnosis and must be present at high titre
- Raynaud's phenomenon is present in 95% and is often the initial manifestation
- Pulmonary arterial hypertension affects 10-25% and is the leading cause of mortality
- 10-year survival is 80-90%
- 30% evolve into a definite connective tissue disease over time
- Annual screening with echocardiography and pulmonary function tests is essential for early PAH detection
- Management is symptom-directed and organ-based, drawing from SLE, SSc, and PM management strategies
Management Approach: "Treatment is tailored to organ manifestations. For Raynaud's, I would use calcium channel blockers first-line. For arthritis, NSAIDs and hydroxychloroquine. For myositis, high-dose corticosteroids with early steroid-sparing agents like methotrexate or mycophenolate. For ILD, I would use mycophenolate or cyclophosphamide depending on severity. PAH requires specialist management with combination therapy including PDE5 inhibitors, endothelin receptor antagonists, and prostanoids. The key is annual screening for PAH and aggressive treatment of myositis and ILD to prevent irreversible damage."
Common Mistakes
❌ Mistakes that fail candidates:
- Failing to mention anti-U1 RNP antibodies as essential for diagnosis
- Not emphasizing annual PAH screening as critical for reducing mortality
- Confusing MCTD with SLE (renal and CNS disease rare in MCTD)
- Suggesting "benign" prognosis (outdated view from Sharp's original description)
- Not recognizing that 30% evolve to definite CTD
- Recommending single-agent therapy for PAH (combination therapy is standard for moderate-severe disease)
- Forgetting that myositis in MCTD generally responds well to corticosteroids
Model Answer: Management Approach
Q: "A 35-year-old woman presents with Raynaud's phenomenon, swollen hands, proximal muscle weakness, and dyspnoea. ANA is positive with a speckled pattern, and anti-U1 RNP is strongly positive. How would you manage this patient?"
A: "This presentation is highly suggestive of mixed connective tissue disease given the combination of Raynaud's, swollen hands, myositis features, and high-titre anti-U1 RNP antibodies. My approach would be systematic and organ-based.
First, I would complete the diagnostic workup to confirm the diagnosis and assess disease extent: full autoimmune serology to exclude other CTDs, muscle enzymes (CK, aldolase), pulmonary function tests with DLCO, transthoracic echocardiography to screen for PAH, and HRCT chest given the dyspnoea. If CK is elevated, I would perform EMG and consider muscle MRI or biopsy to confirm myositis.
The dyspnoea requires urgent investigation to differentiate PAH, ILD, myositis affecting respiratory muscles, or cardiac involvement. PFTs will help: isolated DLCO reduction suggests PAH, whereas a restrictive pattern suggests ILD. Echo will estimate RVSP, and if > 40 mmHg, I would refer for right heart catheterization.
For the myositis, I would initiate high-dose corticosteroids (prednisolone 1 mg/kg/day) and a steroid-sparing agent such as methotrexate 15-25 mg weekly or mycophenolate mofetil 2-3 g/day. I would monitor CK levels weekly initially to assess response.
For the Raynaud's phenomenon, I would start a calcium channel blocker such as nifedipine extended-release 30-60 mg/day and advise smoking cessation and cold avoidance.
If PAH is confirmed, I would refer urgently to a pulmonary hypertension specialist for PAH-specific therapies, likely starting with a PDE5 inhibitor and endothelin receptor antagonist combination. If ILD is present, mycophenolate would treat both myositis and ILD.
Long-term, I would arrange multidisciplinary follow-up with annual echo and PFTs for PAH surveillance, as PAH is the leading cause of mortality in MCTD. I would educate the patient about disease evolution, as 30% progress to definite CTD, and monitor for development of features suggestive of SLE or scleroderma."
References
-
Venables PJW. Mixed connective tissue disease. Lupus. 2006;15(3):132-7. doi:10.1191/0961203306lu2283rr
-
Sharp GC, Irvin WS, Tan EM, et al. Mixed connective tissue disease—an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972;52(2):148-59. doi:10.1016/0002-9343(72)90064-2
-
Hajas A, Szodoray P, Nakken B, et al. Clinical course, prognosis, and causes of death in mixed connective tissue disease. J Rheumatol. 2013;40(7):1134-42. doi:10.3899/jrheum.121272
-
Alarcon-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol. 1989;16(3):328-34. PMID: 2724251
-
Tani C, Carli L, Vagnani S, et al. The diagnosis and classification of mixed connective tissue disease. J Autoimmun. 2014;48-49:46-9. doi:10.1016/j.jaut.2014.01.008
-
Mosca M, Neri R, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): a review of the literature and a proposal for preliminary classification criteria. Clin Exp Rheumatol. 1999;17(5):615-20. PMID: 10544849
-
Burdt MA, Hoffman RW, Deutscher SL, et al. Long-term outcome in mixed connective tissue disease: longitudinal clinical and serologic findings. Arthritis Rheum. 1999;42(5):899-909. doi:10.1002/1529-0131(199905)42:5less than 899::AID-ANR8> 3.0.CO;2-L
-
Ungprasert P, Crowson CS, Chowdhary VR, et al. Epidemiology of mixed connective tissue disease, 1985-2014: a population-based study. Arthritis Care Res (Hoboken). 2016;68(12):1843-8. doi:10.1002/acr.22872
-
Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76(12):1955-64. doi:10.1136/annrheumdis-2017-211468
-
Cappelli S, Randone SB, Camiciottoli G, et al. Interstitial lung disease in systemic sclerosis: where do we stand? Eur Respir Rev. 2015;24(137):411-9. doi:10.1183/16000617.00002915
-
Gunnarsson R, Aaløkken TM, Molberg Ø, et al. Prevalence and severity of interstitial lung disease in mixed connective tissue disease: a nationwide, cross-sectional study. Ann Rheum Dis. 2012;71(12):1966-72. doi:10.1136/annrheumdis-2011-201253
-
Paradowska-Gorycka A, Stypinska B, Olesinska M, et al. Association of HLA-DRB1 alleles with susceptibility to mixed connective tissue disease in Polish patients. HLA. 2016;87(1):13-8. doi:10.1111/tan.12705
-
Swanton J, Isenberg D. Mixed connective tissue disease: still crazy after all these years. Rheum Dis Clin North Am. 2005;31(3):421-36. doi:10.1016/j.rdc.2005.04.009
-
Greidinger EL, Hoffman RW. The appearance of U1 RNP antibody specificities in sequential autoimmune human antisera follows a characteristic order that implicates the U1-70 kDa and B'/B proteins as predominant U1 RNP immunogens. Arthritis Rheum. 2001;44(2):368-75. doi:10.1002/1529-0131(200102)44:2less than 368::AID-ANR53> 3.0.CO;2-5
-
Kattah NH, Kattah MG, Utz PJ. The U1-snRNP complex: structural properties relating to autoimmune pathogenesis in rheumatic diseases. Immunol Rev. 2010;233(1):126-45. doi:10.1111/j.0105-2896.2009.00863.x
-
Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179(4):1317-30. doi:10.1084/jem.179.4.1317
-
Bae SC, Lee YH. Associations between interferon regulatory factor 5 (IRF5) polymorphisms and susceptibility to systemic lupus erythematosus and rheumatoid arthritis: a meta-analysis. Autoimmunity. 2016;49(6):438-45. doi:10.1080/08916934.2016.1181698
-
Tanaka Y, Kuwana M. Pulmonary involvement in mixed connective tissue disease: a comparison with other collagen vascular diseases using high resolution CT. J Comput Assist Tomogr. 2002;26(3):349-57. doi:10.1097/00004728-200205000-00008
-
Lundberg IE. The physiology of inflammatory myopathies: an overview. Acta Physiol Scand. 2001;171(3):207-13. doi:10.1046/j.1365-201x.2001.00829.x
-
Kitridou RC, Akmal M, Turkel SB, et al. Renal involvement in mixed connective tissue disease: a longitudinal clinicopathologic study. Semin Arthritis Rheum. 1986;16(2):135-45. doi:10.1016/0049-0172(86)90049-9
-
Herrick AL. Raynaud's phenomenon and systemic sclerosis. Rheumatology (Oxford). 2005;44(Suppl 3):iii19-iii23. doi:10.1093/rheumatology/kei021
-
Bennett RM, O'Connell DJ. Mixed connective tissue disease: a clinicopathologic study of 20 cases. Semin Arthritis Rheum. 1980;10(1):25-51. doi:10.1016/0049-0172(80)90013-x
-
van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737-47. doi:10.1002/art.38098
-
Kasukawa R, Tojo T, Miyawaki S. Preliminary diagnostic criteria for classification of mixed connective tissue disease. In: Kasukawa R, Sharp GC, eds. Mixed Connective Tissue Disease and Anti-nuclear Antibodies. Amsterdam: Elsevier; 1987:41-7.
-
Ventura F, Canas da Silva P, Alves JD. Pregnancy and mixed connective tissue disease: a systematic review. Clin Rheumatol. 2021;40(7):2609-17. doi:10.1007/s10067-021-05600-9
Last Updated: 2026-01-06
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Systemic Lupus Erythematosus
- Systemic Sclerosis
- Polymyositis and Dermatomyositis
Differentials
Competing diagnoses and look-alikes to compare.
- Undifferentiated Connective Tissue Disease
- Overlap Syndromes
Consequences
Complications and downstream problems to keep in mind.
- Pulmonary Arterial Hypertension
- Interstitial Lung Disease