Primary Biliary Cholangitis
Primary biliary cholangitis (PBC) is a chronic, progressive autoimmune cholestatic liver disease characterised by immune... MRCP exam preparation.
What matters first
Primary biliary cholangitis (PBC) is a chronic, progressive autoimmune cholestatic liver disease characterised by immune... MRCP exam preparation.
Decompensated cirrhosis
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Decompensated cirrhosis
- Inadequate UDCA response
- Rapidly progressive disease
- Variceal bleeding
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- Primary Sclerosing Cholangitis
- Autoimmune Hepatitis
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Primary Biliary Cholangitis
1. Overview
Primary biliary cholangitis (PBC) is a chronic, progressive autoimmune cholestatic liver disease characterised by immune-mediated destruction of small and medium-sized intrahepatic bile ducts. The condition predominantly affects middle-aged women and represents one of the most common causes of chronic cholestatic liver disease. The hallmark laboratory finding is the presence of anti-mitochondrial antibodies (AMA), which are detected in approximately 95% of patients and demonstrate 98% specificity for the diagnosis. [1,2]
The clinical course of PBC varies considerably, ranging from asymptomatic biochemical abnormalities detected incidentally to progressive disease leading to cirrhosis, portal hypertension, and liver failure. Early diagnosis and initiation of treatment with ursodeoxycholic acid (UDCA) can significantly improve outcomes, with many patients achieving normal or near-normal life expectancy. However, approximately 30-40% of patients demonstrate inadequate response to UDCA, requiring second-line therapeutic options. [3,4]
The disease typically presents with fatigue and pruritus, though up to 60% of newly diagnosed patients are asymptomatic at presentation due to increased detection through routine biochemical screening. Without treatment, PBC progresses through histological stages from portal inflammation to fibrosis and ultimately cirrhosis, with complications including portal hypertension, hepatocellular carcinoma, and metabolic bone disease. Understanding response criteria, staging systems, and transplantation thresholds is essential for optimal management. [5,6]
2. Epidemiology
PBC demonstrates distinctive epidemiological features with marked female predominance and geographic variation in incidence and prevalence.
Incidence and Prevalence
The annual incidence of PBC ranges from 0.3 to 5.8 cases per 100,000 population, with prevalence estimates between 1.9 and 40.2 per 100,000. [7] The highest prevalence rates are reported in Northern Europe and North America, suggesting both genetic and environmental influences on disease susceptibility. Population-based studies indicate increasing detection rates over recent decades, likely reflecting improved awareness and more widespread use of liver function testing. [1]
| Statistic | Value | Notes |
|---|---|---|
| Annual incidence | 0.3-5.8 per 100,000 | Higher in Northern Europe |
| Prevalence | 1.9-40.2 per 100,000 | Geographic variation |
| Female:Male ratio | 9:1 to 10:1 | Marked female predominance |
| Peak age at diagnosis | 40-60 years | Can occur at any age |
| Family history | 1-6% | First-degree relatives affected |
Demographics
PBC predominantly affects women, with female:male ratios ranging from 9:1 to 10:1. [2] The peak age at diagnosis is between 40 and 60 years, though the disease can present across a wide age spectrum. Men tend to present at older ages and may have more advanced disease at diagnosis. [8]
Risk Factors
Several epidemiological associations have been identified:
-
Genetic factors: First-degree relatives of PBC patients have a prevalence approximately 1000-fold higher than the general population, with concordance rates of 1-6%. [9] Genome-wide association studies have identified multiple susceptibility loci, particularly in the HLA region and genes involved in immune regulation.
-
Environmental factors: Smoking has been associated with increased risk, with odds ratios of 1.5-2.0 in several studies. [10] Urinary tract infections and other bacterial infections have been implicated as potential triggers through molecular mimicry mechanisms.
-
Geographic clustering: Higher prevalence in northern latitudes suggests environmental or genetic factors specific to these populations.
Trends
The apparent increase in PBC incidence over recent decades likely reflects improved case ascertainment rather than true increasing incidence. Earlier diagnosis in the asymptomatic phase has become more common with routine biochemical screening. [1]
3. Aetiology and Pathophysiology
Aetiology
PBC is an autoimmune disease of unknown aetiology, though multiple factors contribute to disease development:
Primary mechanisms:
- Breakdown of immune tolerance to mitochondrial antigens, specifically the E2 component of pyruvate dehydrogenase complex (PDC-E2)
- Genetic susceptibility involving HLA and non-HLA immune-related genes
- Environmental triggers potentially including infectious agents or xenobiotics with molecular mimicry to self-antigens
Associated conditions: PBC frequently coexists with other autoimmune diseases, supporting a shared immunopathogenic basis. These include:
- Sjögren syndrome (up to 70% of PBC patients) [11]
- Autoimmune thyroid disease (20-25%)
- Systemic sclerosis (7-12%)
- Rheumatoid arthritis
- Raynaud phenomenon
Pathophysiology
Molecular Pathogenesis:
The fundamental pathological process in PBC involves immune-mediated destruction of small intrahepatic bile ducts (40-80 μm diameter). The primary autoantigen is the E2 subunit of the pyruvate dehydrogenase complex (PDC-E2), a mitochondrial enzyme located on the inner mitochondrial membrane. [12]
Immunological Cascade:
-
Initiation: Loss of tolerance to PDC-E2 through mechanisms that may include:
- Molecular mimicry following bacterial infection
- Aberrant apoptosis of biliary epithelial cells with membrane blebs containing intact PDC-E2
- Genetic predisposition reducing immune tolerance thresholds
-
Amplification: CD4+ and CD8+ T cells infiltrate portal tracts, targeting biliary epithelial cells expressing PDC-E2 on their surface. B cells produce anti-mitochondrial antibodies that may contribute to epithelial damage through complement-dependent mechanisms or antibody-dependent cellular cytotoxicity.
-
Progression: Ongoing inflammation leads to:
- Bile duct destruction and ductopenia (reduction in number of bile ducts)
- Cholestasis with accumulation of toxic bile acids
- Secondary injury from bile acid-mediated hepatocyte damage
- Progressive fibrosis and cirrhosis development
Histological Evolution:
The Scheuer staging system describes four histological stages: [13]
- Stage I (Portal stage): Portal inflammation with florid duct lesions - granulomatous destruction of interlobular bile ducts with lymphocytic infiltration and epithelioid granulomas
- Stage II (Periportal stage): Periportal fibrosis with interface hepatitis, bile duct proliferation
- Stage III (Septal stage): Bridging fibrosis with architectural distortion
- Stage IV (Cirrhotic stage): Established cirrhosis with regenerative nodules
The Ludwig classification is similar but emphasizes different features. Both systems correlate with prognosis, though significant sampling variability limits clinical utility in individual patients.
Cholestatic Injury Mechanisms:
Accumulation of toxic hydrophobic bile acids (chenodeoxycholic acid, deoxycholic acid) causes:
- Mitochondrial dysfunction in hepatocytes
- Oxidative stress and lipid peroxidation
- Activation of apoptotic pathways
- Activation of hepatic stellate cells leading to fibrogenesis
UDCA therapy works by replacing toxic endogenous bile acids with the non-toxic, hydrophilic UDCA, reducing hepatocellular injury and potentially exerting immunomodulatory and anti-apoptotic effects. [14]
4. Clinical Presentation
Symptoms
The clinical spectrum of PBC has evolved, with many patients now diagnosed in the asymptomatic phase through incidental laboratory findings.
Cardinal symptoms:
-
Fatigue (20-70% of patients): Often the most debilitating symptom, characterised by profound tiredness disproportionate to disease activity. The pathophysiology remains unclear but may involve central nervous system effects of accumulated bile acids or cytokines. Fatigue severity does not correlate with disease stage or liver biochemistry. [15]
-
Pruritus (20-70% at presentation): Typically worse at night, exacerbated by warmth, and affecting palms and soles initially before becoming generalized. Results from accumulation of pruritogens (bile acids, lysophosphatidic acid, autotaxin). Can precede other manifestations by months to years. In severe cases, pruritus profoundly impacts quality of life and may be an indication for liver transplantation. [5]
-
Asymptomatic (up to 60% at diagnosis): Detected through incidental finding of elevated alkaline phosphatase on routine blood tests. These patients may still experience reduced quality of life despite absence of classic symptoms.
Associated symptoms:
- Right upper quadrant discomfort (uncommon, non-specific)
- Symptoms of complications: jaundice, ascites, encephalopathy (late disease)
- Sicca symptoms (dry eyes, dry mouth) reflecting associated Sjögren syndrome
- Metabolic bone disease symptoms (bone pain, fractures)
Red flag symptoms:
- Jaundice (indicates advanced disease or decompensation)
- Variceal bleeding (portal hypertension)
- Progressive weight loss
- Severe refractory pruritus
| Symptom | Frequency at Presentation | Clinical Significance |
|---|---|---|
| Asymptomatic | 50-60% | Early detection, better prognosis |
| Fatigue | 20-70% | Major quality of life impact, no specific treatment |
| Pruritus | 20-70% | May precede diagnosis, can be intractable |
| Jaundice | less than 10% initially | Indicates advanced disease, poor prognosis |
Signs
Early disease: Most patients with early PBC have no specific physical findings. Some may exhibit:
- Xanthelasma (cholesterol deposits around eyelids, 10-25%)
- Xanthomata (on extensor surfaces, elbows, knees - rare, indicates prolonged hypercholesterolaemia)
- Excoriation marks from scratching (chronic pruritus)
- Features of associated conditions (dry eyes/mouth, Raynaud phenomenon, sclerodactyly)
Intermediate disease:
- Hepatomegaly (may be smooth and non-tender)
- Mild splenomegaly (developing portal hypertension)
- Hyperpigmentation (melanin deposition from chronic cholestasis)
Advanced disease (cirrhosis and portal hypertension):
- Jaundice
- Spider naevi
- Palmar erythema
- Ascites
- Peripheral oedema
- Asterixis (hepatic encephalopathy)
- Splenomegaly
- Caput medusae (rare)
| Finding | Stage | Prevalence | Significance |
|---|---|---|---|
| Xanthelasma | Early-intermediate | 10-25% | Chronic cholestasis, hypercholesterolaemia |
| Hepatomegaly | Any | 25-50% | Non-specific |
| Splenomegaly | Advanced | 30-50% | Portal hypertension |
| Jaundice | Advanced | Variable | Poor prognostic sign |
| Ascites | Decompensated | Variable | Requires transplant evaluation |
5. Differential Diagnosis
The differential diagnosis of chronic cholestatic liver disease is crucial, as management and prognosis differ significantly between conditions.
Primary Differentials
-
Primary Sclerosing Cholangitis (PSC)
- Key differences: Male predominance, strong association with inflammatory bowel disease (70-80%), affects large and medium bile ducts
- AMA negative
- MRCP shows multifocal strictures and beading of bile ducts
- p-ANCA positive in 60-80%
-
Autoimmune Hepatitis (AIH)
- Hepatitic rather than cholestatic pattern (elevated ALT/AST predominates)
- ANA, anti-smooth muscle antibodies positive
- Higher IgG rather than IgM
- May overlap with PBC (10-15% of cases have "overlap syndrome")
-
Drug-Induced Liver Injury (Cholestatic pattern)
- Temporal relationship with medication use
- Common culprits: chlorpromazine, azathioprine, amoxicillin-clavulanate
- AMA negative
- Improvement on drug cessation
-
Sarcoidosis
- Systemic features: lymphadenopathy, pulmonary involvement, hypercalcaemia
- Granulomas on liver biopsy (non-caseating, without bile duct damage)
- AMA negative
- Elevated ACE levels
-
AMA-Negative PBC
- 5% of PBC patients lack AMA but have identical clinical, biochemical, and histological features
- Often have anti-nuclear antibodies (anti-sp100, anti-gp210)
- Liver biopsy diagnostic showing florid duct lesions
-
Chronic Biliary Obstruction
- History of biliary surgery, gallstones, malignancy
- Imaging (ultrasound, MRCP) shows dilated bile ducts
- AMA negative
| Differential | Key Distinguishing Features | Diagnostic Test |
|---|---|---|
| PSC | Male, IBD association, large duct involvement | MRCP showing strictures |
| AIH | Hepatitic pattern, elevated IgG, ANA/ASMA+ | Liver biopsy, autoantibodies |
| Drug-induced | Temporal relationship, drug exposure | History, exclusion |
| Sarcoidosis | Systemic features, ACE elevated | Biopsy showing non-caseating granulomas |
| AMA-negative PBC | Cholestatic, anti-sp100/gp210 positive | Liver biopsy |
| Biliary obstruction | Dilated ducts on imaging | Ultrasound/MRCP |
PBC-AIH Overlap Syndrome
Approximately 10% of patients demonstrate features of both PBC and AIH. The Paris criteria define overlap as:
PBC features (2 of 3):
- ALP ≥2× ULN or GGT ≥5× ULN
- AMA positive
- Florid duct lesion on biopsy
AIH features (2 of 3):
- ALT ≥5× ULN
- IgG ≥2× ULN or ASMA positive
- Interface hepatitis on biopsy
Overlap syndrome requires treatment with both UDCA and immunosuppression (prednisolone ± azathioprine). [16]
6. Investigations
Diagnosis of PBC relies on characteristic biochemical abnormalities, serological markers, and, when necessary, histological findings.
First-Line Investigations
Liver biochemistry:
- Alkaline phosphatase (ALP): Elevated, often 2-10× upper limit of normal (ULN), cholestatic pattern
- Gamma-glutamyl transferase (GGT): Usually elevated parallel to ALP
- Aminotransferases (ALT/AST): Normal to mildly elevated (less than 5× ULN); higher elevation suggests overlap syndrome
- Bilirubin: Normal in early disease; progressive elevation indicates advanced disease and poor prognosis
- Albumin: Preserved until advanced disease
- Prothrombin time/INR: Prolonged in advanced disease or vitamin K deficiency
Serological markers:
- Anti-mitochondrial antibodies (AMA): Positive (titre ≥1:40) in 95% of patients. AMA-M2 (specific for PDC-E2) has 95% sensitivity and 98% specificity. [1,17]
- Immunoglobulins: IgM typically elevated (70-80% of cases), a distinguishing feature from other liver diseases
- Other autoantibodies: ANA (30-50%, often speckled pattern with anti-sp100 or anti-gp210 specificity in AMA-negative cases)
Imaging:
- Liver ultrasound: Exclude biliary obstruction, assess for cirrhosis, screen for HCC in established cirrhosis
- Transient elastography (FibroScan): Non-invasive assessment of liver stiffness to estimate fibrosis stage
Second-Line Investigations
Liver biopsy: Not routinely required for diagnosis when AMA-positive with cholestatic biochemistry. Indications include:
- AMA-negative cholestatic liver disease
- Atypical features suggesting overlap syndrome or alternative diagnosis
- Assessment of fibrosis stage when non-invasive methods inconclusive
Histological features:
- Stage I: Florid duct lesion - granulomatous destruction of bile ducts with lymphocytic infiltration
- Stage II: Periportal inflammation and fibrosis
- Stage III: Bridging fibrosis
- Stage IV: Cirrhosis
MRCP (Magnetic Resonance Cholangiopancreatography):
- Indicated when diagnosis uncertain to exclude PSC or biliary obstruction
- Normal in PBC (small ducts not visualized)
Screening for Complications and Associated Conditions
- Bone density (DEXA scan): Screen for osteoporosis at diagnosis and monitor (cholestasis increases fracture risk)
- Fat-soluble vitamin levels: Vitamins A, D, E, K in patients with jaundice or malabsorption
- Lipid profile: Hypercholesterolaemia common but cardiovascular risk paradoxically not increased
- Thyroid function: Screen for autoimmune thyroid disease
- Schirmer test, salivary flow: If sicca symptoms suggest Sjögren syndrome
Diagnostic Criteria
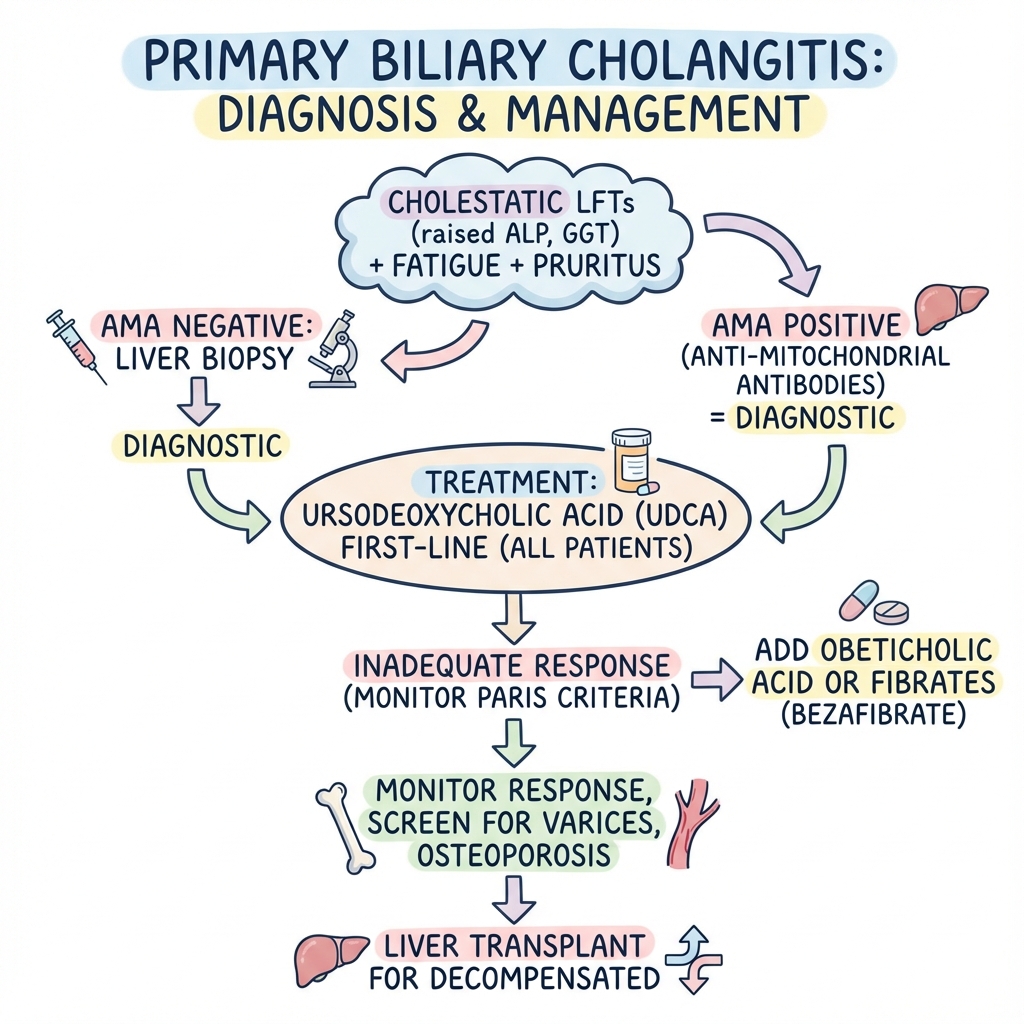
PBC is diagnosed when 2 of the following 3 criteria are met (EASL guidelines): [1]
- Cholestatic liver biochemistry (elevated ALP)
- Positive AMA (or specific anti-nuclear antibodies if AMA-negative)
- Liver biopsy showing characteristic histology
| Investigation | Typical Finding | Sensitivity | Specificity |
|---|---|---|---|
| AMA (M2) | Positive (≥1:40) | 95% | 98% |
| ALP | Elevated (≥1.5× ULN) | > 95% | Variable |
| IgM | Elevated | 70-80% | Low |
| Liver biopsy | Florid duct lesion (Stage I) | Gold standard | High for PBC |
Interpretation Pearls:
-
AMA subtypes: M2 (PDC-E2) is most specific for PBC. Other mitochondrial antigens (M4, M8, M9) less specific.
-
AMA-negative PBC: 5% lack AMA. Look for anti-sp100 (20-30% of AMA-neg PBC) or anti-gp210 (10-40% of AMA-neg PBC). Anti-gp210 positivity associated with more aggressive disease and higher risk of progression to liver failure.
-
Bilirubin prognostic importance: Normal bilirubin at presentation and after UDCA treatment indicates good prognosis. Progressive bilirubin elevation is strongest predictor of mortality and need for transplantation.
-
Transient elastography thresholds: Liver stiffness less than 9 kPa suggests minimal fibrosis; > 17 kPa suggests cirrhosis. Useful for monitoring progression and stratifying surveillance needs.
-
Biopsy sampling error: Portal tracts may be unevenly affected; florid duct lesions may not be present in all samples. Stage assigned is based on most advanced area observed.
7. Classification and Staging
Histological Staging Systems
Two main systems classify PBC histologically:
Scheuer Staging (1967): [13]
- Stage I: Portal inflammation, florid duct lesions
- Stage II: Periportal inflammation, bile duct proliferation
- Stage III: Septal fibrosis, bridging
- Stage IV: Cirrhosis
Ludwig Staging (1978): Similar four-stage system emphasizing portal inflammation, periportal changes, septal fibrosis, and cirrhosis.
Both correlate with prognosis but have limitations due to sampling variability and patchy disease distribution. Non-invasive methods (transient elastography) increasingly used for fibrosis assessment.
Biochemical Response Criteria
Multiple criteria assess response to UDCA therapy, predicting long-term outcomes:
Paris-I Criteria (1 year after UDCA): [18]
- ALP less than 3× ULN
- AST less than 2× ULN
- Bilirubin normal
Meeting all three criteria predicts excellent long-term survival similar to age-matched controls.
Barcelona Criteria (1 year):
- ALP decrease ≥40% of baseline OR ALP normal
UK-PBC Score:
- Risk stratification tool incorporating baseline albumin, bilirubin, ALP, platelets, and transaminases
- Provides individualised 5, 10, and 15-year survival estimates
GLOBE Score:
- International score using age, bilirubin, ALP, albumin, platelets
- Validated across multiple cohorts for transplant-free survival prediction
Response criteria identify patients requiring second-line therapy. Approximately 30-40% of UDCA-treated patients fail to achieve biochemical response and have increased risk of progression. [3]
| Criteria | Definition | Predictive Value |
|---|---|---|
| Paris-I | ALP less than 3×, AST less than 2×, bili normal @ 1y | High: survival comparable to general population |
| Barcelona | ALP decrease ≥40% or normalisation @ 1y | Good: identifies responders |
| UK-PBC score | Multi-variable calculator | Continuous risk score, well-validated |
| GLOBE score | Age, bili, ALP, alb, plt | Transplant-free survival at 5-15 years |
8. Management
Management of PBC aims to slow disease progression, manage symptoms, prevent complications, and identify candidates for liver transplantation.
Diagnostic Algorithm and Initial Management
- Confirm diagnosis (AMA + cholestatic biochemistry ± biopsy)
- Assess disease stage (biochemistry, imaging, elastography)
- Screen for complications (varices if cirrhotic, bone disease)
- Initiate UDCA therapy
- Assess response at 1 year using validated criteria
- Manage symptoms (pruritus, fatigue)
- Monitor for progression and complications
First-Line Medical Therapy
Ursodeoxycholic Acid (UDCA):
UDCA is the only approved first-line therapy for PBC and should be initiated in all patients regardless of symptoms or disease stage. [1]
- Dose: 13-15 mg/kg/day orally in 2-3 divided doses (with meals to enhance absorption)
- Mechanism: Replaces toxic endogenous bile acids with cytoprotective UDCA; stimulates bile flow; immunomodulatory effects
- Efficacy: Improves liver biochemistry in 60-70%; reduces progression to cirrhosis and need for transplantation; improves survival in responders to levels approaching general population [14]
- Safety: Excellent safety profile; rarely causes diarrhea (reduce dose or switch to once-daily dosing)
- Duration: Lifelong therapy
Evidence base: Multiple randomized controlled trials and meta-analyses demonstrate UDCA efficacy in slowing histological progression and improving transplant-free survival, particularly in patients with early disease and those achieving biochemical response. [14]
Second-Line Therapies (Inadequate UDCA Response)
For patients who fail to achieve adequate biochemical response to UDCA (30-40%), second-line options include:
Obeticholic Acid (OCA):
- Mechanism: Farnesoid X receptor (FXR) agonist; reduces bile acid synthesis and enhances bile acid transport
- Indication: Inadequate response or intolerance to UDCA
- Dose: Initial 5 mg once daily; increase to 10 mg daily after 3-6 months based on tolerability and response
- Efficacy: POISE trial demonstrated 46% of patients achieved ALP less than 1.67× ULN, total bilirubin ≤ULN, and ≥15% ALP reduction vs 10% placebo [19]
- Adverse effects: Pruritus (most common, dose-related; occurs in 50-70%); may require dose reduction
- Contraindications: Decompensated cirrhosis (Child-Pugh B or C) - risk of worsening hepatic function
- Cost: Expensive; access varies by healthcare system
Fibrates (Bezafibrate, Fenofibrate):
- Mechanism: PPAR-α agonists; unclear exact mechanism in PBC but improve cholestasis and may reduce inflammation
- Evidence: BEZURSO trial showed bezafibrate + UDCA superior to UDCA alone for achieving biochemical response [20]
- Dose: Bezafibrate 400 mg daily (reduce in renal impairment); fenofibrate 200 mg daily
- Efficacy: 60-80% achieve biochemical response; particularly effective for pruritus
- Safety: Generally well-tolerated; monitor creatinine (may cause reversible increase); risk of myopathy (especially with statins)
- Availability: Off-label use; not approved by FDA for PBC but used in Europe
Budesonide:
- Limited role: May be considered in overlap syndrome with AIH features
- Contraindication: Cirrhosis with portal hypertension (risk of portal vein thrombosis)
| Treatment | Indication | Dose | Response Rate | Key Adverse Effect |
|---|---|---|---|---|
| UDCA | First-line, all patients | 13-15 mg/kg/day | 60-70% | Diarrhea (rare) |
| Obeticholic acid | Second-line, UDCA inadequate response | 5-10 mg daily | 25-30% additional response | Pruritus (50-70%) |
| Bezafibrate | Second-line, off-label | 400 mg daily | 60-80% when added to UDCA | Myalgia, creatinine elevation |
| Fenofibrate | Second-line, off-label | 200 mg daily | Similar to bezafibrate | Similar to bezafibrate |
Symptom Management
Pruritus:
Pruritus can be debilitating and refractory to treatment. Stepwise approach:
-
First-line: Cholestyramine (bile acid sequestrant)
- Dose: 4-16 g/day in divided doses, taken 4 hours apart from other medications
- Efficacy: Effective in 80-90% with mild-moderate pruritus
- Adverse effects: Constipation, bloating; may impair absorption of fat-soluble vitamins and other drugs
-
Second-line: Rifampicin
- Dose: 150-300 mg daily, increase to 600 mg if needed
- Mechanism: Induces cytochrome P450 enzymes, enhancing bile acid metabolism
- Efficacy: 50-60% experience relief
- Adverse effects: Hepatotoxicity (monitor LFTs closely); drug interactions (CYP inducer)
-
Third-line: Naltrexone/Naloxone
- Opioid antagonists; mechanism unclear but may relate to endogenous opioids
- Dose: Naltrexone 12.5-50 mg daily
- Start low and titrate to minimize opioid withdrawal-like symptoms
- Efficacy: Variable
-
Other options:
- Sertraline (SSRI): 75-100 mg daily; modest benefit
- Antihistamines: Limited efficacy; may help nocturnal pruritus through sedation
- UV phototherapy: May provide temporary relief
-
Refractory pruritus:
- Indication for liver transplantation if severely impacting quality of life
- Plasmapheresis, molecular adsorbent recirculating system (MARS): Temporary bridge to transplant
Fatigue:
No effective pharmacological treatment exists for PBC-associated fatigue. Management focuses on:
- Excluding other causes (hypothyroidism, anaemia, depression, sleep disorders)
- Sleep hygiene optimization
- Graded exercise programs
- Psychological support
- Modafinil: Small studies show no consistent benefit
Sicca symptoms:
For associated Sjögren syndrome:
- Artificial tears for dry eyes
- Saliva substitutes, sugar-free gum for dry mouth
- Pilocarpine or cevimeline for severe sicca (muscarinic agonists)
Monitoring and Follow-Up
Frequency:
- Every 6-12 months for stable disease
- Every 3-6 months for inadequate treatment response or advanced disease
Monitoring parameters:
- Liver biochemistry (ALP, bilirubin, albumin, AST/ALT)
- Full blood count, platelets (thrombocytopenia suggests portal hypertension)
- Calculate prognostic scores (UK-PBC, GLOBE)
- Transient elastography annually to assess fibrosis progression
Cirrhosis surveillance:
- Upper GI endoscopy at diagnosis of cirrhosis to screen for varices
- Repeat every 2-3 years if no varices; every 1-2 years if small varices
- Ultrasound + AFP every 6 months for HCC surveillance
- DEXA scan for osteoporosis screening
Vaccination:
- Hepatitis A and B vaccines (all patients with chronic liver disease)
- Pneumococcal, influenza vaccines annually
- COVID-19 vaccination
Management of Complications
Portal hypertension:
- Non-selective beta-blockers (propranolol, carvedilol) for variceal bleeding prophylaxis
- Endoscopic variceal ligation for large varices or bleeding
- TIPS (transjugular intrahepatic portosystemic shunt) for refractory ascites or variceal bleeding
Metabolic bone disease:
- Calcium 1000-1500 mg daily + Vitamin D 800-1000 IU daily
- Bisphosphonates for osteoporosis
- Avoid prolonged corticosteroids
Fat-soluble vitamin deficiency:
- Supplement vitamins A, D, E, K in patients with jaundice or steatorrhea
- Monitor vitamin levels in advanced disease
Hepatocellular carcinoma:
- Risk 3-4 times higher than general population in cirrhotic PBC
- 6-monthly surveillance with ultrasound ± AFP
Liver Transplantation
Indications:
- Decompensated cirrhosis (ascites, variceal bleeding, encephalopathy)
- Hepatocellular carcinoma (within Milan criteria)
- MELD score ≥15 (allocation based on MELD score in most systems)
- Intractable pruritus despite maximal medical therapy
- Progressive jaundice
Outcomes:
- Excellent: 5-year survival 80-85%, 10-year survival 75%
- PBC may recur in allograft (10-30% at 10 years) but rarely causes graft failure
- Continuation of UDCA post-transplant may reduce recurrence risk
Prognostic Models for Transplant Timing:
Mayo Risk Score (original model): Incorporates age, bilirubin, albumin, prothrombin time, presence of oedema. Less used now due to development of MELD.
MELD Score: Standard allocation score in most transplant systems; calculated from bilirubin, creatinine, INR.
UK-PBC and GLOBE scores: Better for long-term risk prediction in earlier disease; help identify patients who may benefit from more aggressive monitoring or second-line therapy.
Paris Criteria Application:
At 1-year post-UDCA initiation, assess:
- ALP: Must be less than 3× ULN
- AST: Must be less than 2× ULN
- Total bilirubin: Must be within normal range
Example:
- Baseline: ALP 450 U/L (ULN 120), AST 95 U/L (ULN 40), Bili 12 μmol/L (ULN 21)
- 1 year on UDCA: ALP 280 U/L, AST 65 U/L, Bili 15 μmol/L
- Evaluation: ALP 280/120 = 2.3× (meets criterion less than 3×); AST 65/40 = 1.6× (meets criterion less than 2×); Bili normal (meets criterion)
- Conclusion: Adequate response; continue UDCA monotherapy; good long-term prognosis
Non-responder would have ALP ≥3× ULN, AST ≥2× ULN, or elevated bilirubin. Consider adding obeticholic acid or fibrate.
Obeticholic Acid Dosing Adjustments:
- Child-Pugh Class A: Start 5 mg daily; may increase to 10 mg after 3 months if tolerated
- Child-Pugh Class B or C: Contraindicated - reports of hepatic decompensation and death
- Pruritus management: If intolerable pruritus develops, reduce to 5 mg once or twice weekly rather than discontinuing
Transplant Criteria (General):
Refer for transplant evaluation when:
- MELD ≥15
- Development of decompensation (first episode of ascites, variceal bleeding, encephalopathy)
- HCC within transplantable criteria
- Intractable pruritus unresponsive to all medical therapies significantly impairing quality of life
- Progressive cholestasis with bilirubin > 100 μmol/L (6 mg/dL)
9. Complications
Progressive PBC leads to multiple hepatic and extra-hepatic complications:
Hepatic Complications
| Complication | Incidence | Pathophysiology | Management |
|---|---|---|---|
| Cirrhosis | 30-40% over 10-20 years (untreated) | Progressive fibrosis from chronic inflammation | UDCA slows progression; transplant for decompensation |
| Portal hypertension | ~40% in cirrhotic patients | Architectural distortion, increased intrahepatic resistance | Beta-blockers, endoscopic therapy, TIPS |
| Variceal bleeding | 20-30% of cirrhotic patients | Portal hypertension | Primary/secondary prophylaxis with beta-blockers, EVL |
| Ascites | Variable in decompensated disease | Portal hypertension, hypoalbuminaemia | Sodium restriction, diuretics, paracentesis |
| Hepatic encephalopathy | Variable in decompensated disease | Portosystemic shunting, hepatocyte dysfunction | Lactulose, rifaximin |
| Hepatocellular carcinoma | 5-10% cumulative incidence in cirrhosis | Chronic inflammation, cirrhosis | 6-monthly surveillance, transplant if within criteria |
Extra-Hepatic Complications
Metabolic bone disease (40-50%):
- Osteoporosis from vitamin D malabsorption, cholestasis effects on bone metabolism
- Increased fracture risk (relative risk 1.9-2.3)
- Prevention: Calcium, vitamin D supplementation; bisphosphonates if osteoporotic
Fat-soluble vitamin deficiency:
- Vitamins A, D, E, K malabsorption in cholestasis/cirrhosis
- Consequences: Night blindness (A), bone disease (D), neuropathy (E), coagulopathy (K)
- Monitor and supplement as needed
Hyperlipidaemia (70-80%):
- Elevated total and LDL cholesterol, often marked
- Paradoxically, cardiovascular risk not increased in PBC (lipoprotein X accumulation, not atherogenic)
- Statins safe if needed for other indications; not routinely required
Sicca syndrome (Sjögren association):
- Dry eyes (keratoconjunctivitis sicca)
- Dry mouth (xerostomia), increased dental caries risk
- Vaginal dryness
Thyroid disease (20-25%):
- Autoimmune hypothyroidism or hyperthyroidism
- Screen thyroid function periodically
Other autoimmune associations:
- Systemic sclerosis (7-12%), particularly limited cutaneous form (CREST)
- Raynaud phenomenon
- Rheumatoid arthritis
- Celiac disease
Prognosis
Prognosis depends on disease stage at diagnosis, biochemical response to therapy, and development of complications.
Asymptomatic patients with biochemical response to UDCA:
- Life expectancy approaching that of age-matched general population
- Low risk of progression to cirrhosis
Symptomatic or inadequate UDCA response:
- Median survival without transplant 10-15 years
- Risk of progression to cirrhosis and liver-related mortality significantly higher
Advanced disease with elevated bilirubin:
- Bilirubin > 100 μmol/L (6 mg/dL): Median survival 2-3 years without transplant
- Decompensated cirrhosis: Requires urgent transplant evaluation
Prognostic markers (poor prognosis):
- Persistently elevated or rising bilirubin
- Failure to achieve Paris criteria response
- High UK-PBC or GLOBE score
- Presence of cirrhosis at diagnosis
- Anti-gp210 antibody positivity
- Male sex (worse outcomes than females)
- Younger age at presentation (more aggressive disease course)
10. Prevention and Screening
Primary prevention: No established primary prevention strategies exist, as aetiology remains incompletely understood. Avoidance of smoking may reduce risk.
Screening:
- Routine population screening not recommended
- Consider screening first-degree relatives of PBC patients (1000-fold increased risk) if unexplained symptoms or abnormal liver biochemistry
- Screen patients with other autoimmune diseases (particularly Sjögren syndrome, systemic sclerosis) for cholestatic liver biochemistry
Secondary prevention:
- Early initiation of UDCA prevents progression in most patients
- Screening for complications in established disease (varices, HCC, bone disease)
11. Key Guidelines
European Association for the Study of the Liver (EASL) 2017: [1]
- Diagnostic criteria: 2 of 3 (cholestatic biochemistry, AMA+, compatible biopsy)
- UDCA 13-15 mg/kg/day first-line for all patients
- Assess response at 1 year using validated criteria
- Obeticholic acid second-line for inadequate response
American Association for the Study of Liver Diseases (AASLD) 2018:
- Similar diagnostic and treatment recommendations
- Emphasises risk stratification using prognostic scores
British Society of Gastroenterology (BSG):
- Endorses EASL guidelines
- Specific guidance on monitoring and management of complications
12. Examination Focus
Common Exam Questions
- "What are the diagnostic criteria for primary biliary cholangitis?"
- "How would you assess response to ursodeoxycholic acid therapy?"
- "Describe the management of a patient with PBC who has inadequate response to UDCA."
- "What are the indications for liver transplantation in PBC?"
- "How would you investigate a patient with chronic cholestasis?"
- "What is PBC-AIH overlap syndrome and how is it managed?"
- "Describe the pathophysiology of bile duct destruction in PBC."
Viva Points
Opening Statement: "Primary biliary cholangitis is a chronic autoimmune cholestatic liver disease characterised by immune-mediated destruction of small intrahepatic bile ducts, predominantly affecting middle-aged women with a female:male ratio of 9:1."
Key Facts to Mention:
- Anti-mitochondrial antibodies (AMA-M2) are 95% sensitive and 98% specific for diagnosis
- UDCA 13-15 mg/kg/day is first-line therapy and improves outcomes in responders
- Paris criteria assess UDCA response at 1 year: ALP less than 3× ULN, AST less than 2× ULN, bilirubin normal
- 30-40% have inadequate UDCA response requiring second-line therapy (obeticholic acid, fibrates)
- Liver transplantation indicated for decompensated disease with excellent outcomes (5-year survival 80-85%)
Classification Systems Ready to Quote:
- Scheuer/Ludwig histological staging (I-IV: portal inflammation → cirrhosis)
- Paris-I criteria for treatment response
- Mayo Risk Score, MELD score for transplant timing
Evidence to Cite:
- POISE trial (Nevens 2016): Obeticholic acid efficacy in UDCA non-responders
- BEZURSO trial: Bezafibrate + UDCA combination therapy
- EASL 2017 guidelines for diagnostic and management approach
Common Mistakes to Avoid
❌ Diagnostic errors:
- Failing to exclude biliary obstruction with imaging before diagnosing PBC
- Not recognizing AMA-negative PBC (5% of cases)
- Missing PBC-AIH overlap syndrome (needs combined treatment)
❌ Management errors:
- Underdosing UDCA (must use 13-15 mg/kg, not fixed dose)
- Not assessing treatment response at 1 year with validated criteria
- Using obeticholic acid in decompensated cirrhosis (Child-Pugh B/C) - contraindicated
- Failing to screen for varices and HCC in cirrhotic patients
❌ Prognostic errors:
- Not recognizing bilirubin elevation as most important prognostic marker
- Delaying transplant referral until too late in progressive disease
Model Answers
Q: Describe your approach to a 52-year-old woman with elevated ALP found on routine blood tests.
A: "I would approach this systematically. First, I would confirm the ALP elevation and determine if it's hepatic or bony origin by checking GGT - if GGT elevated, likely hepatic. I would take a detailed history for symptoms of liver disease (fatigue, pruritus, jaundice), risk factors for liver disease, medications, and autoimmune disease history.
On examination, I would look for stigmata of chronic liver disease, features of cholestasis (jaundice, xanthelasma, excoriation marks), hepatosplenomegaly, and signs of associated autoimmune conditions.
Initial investigations would include full liver biochemistry, anti-mitochondrial antibodies (AMA-M2), immunoglobulins, and liver ultrasound to exclude biliary obstruction. If AMA positive with cholestatic pattern and no biliary dilatation, the diagnosis is PBC based on EASL criteria (2 of 3: cholestatic biochemistry, AMA+, compatible biopsy).
I would stage the disease with transient elastography and screen for complications including bone density, thyroid function, and if cirrhotic, upper GI endoscopy for varices.
Management would involve initiating UDCA 13-15 mg/kg/day and assessing response at 1 year using Paris criteria. I would provide patient education about the condition, prognosis, and need for lifelong monitoring."
Q: A patient with PBC on UDCA for 1 year has ALP 400 U/L (ULN 120), AST 90 U/L (ULN 40), bilirubin 18 μmol/L (ULN 21). What is your assessment and management?
A: "This patient has inadequate response to UDCA based on Paris criteria. Calculating the multiples of ULN: ALP is 3.3× ULN (criterion failed - requires less than 3×), AST is 2.25× ULN (criterion failed - requires less than 2×), bilirubin is normal (criterion met).
Failing to meet Paris criteria places this patient at increased risk of disease progression and adverse outcomes. I would first verify UDCA compliance and dosing (should be 13-15 mg/kg/day), and exclude other causes of cholestasis or liver injury.
For second-line therapy, I would consider adding obeticholic acid 5 mg daily (FXR agonist), increasing to 10 mg after 3 months if tolerated, based on POISE trial evidence showing improved biochemical response in UDCA non-responders. I would counsel about pruritus risk (50-70%) and ensure no decompensated cirrhosis (contraindication).
Alternative would be off-label fibrate therapy (bezafibrate 400 mg daily or fenofibrate 200 mg daily) based on BEZURSO trial, monitoring renal function.
I would intensify monitoring to 3-6 monthly intervals, calculate prognostic scores (UK-PBC, GLOBE), and ensure surveillance for complications is in place. If Child-Pugh score or MELD score worsens, I would refer for transplant evaluation."
References
-
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145-172. doi:10.1016/j.jhep.2017.03.022
-
Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386(10003):1565-1575. doi:10.1016/S0140-6736(15)00154-3
-
Lammers WJ, van Buuren HR, Hirschfield GM, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147(6):1338-1349. doi:10.1053/j.gastro.2014.08.029
-
Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet. 2013;382(9904):1587-1599. doi:10.1016/S0140-6736(13)60096-3
-
Beuers U, Gershwin ME, Gish RG, et al. Changing nomenclature for PBC: From 'cirrhosis' to 'cholangitis'. Hepatology. 2015;62(5):1620-1622. doi:10.1002/hep.28140
-
Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69(1):394-419. doi:10.1002/hep.30145
-
Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol. 2012;56(5):1181-1188. doi:10.1016/j.jhep.2011.10.025
-
Prince MI, Chetwynd A, Craig WL, Metcalf JV, James OF. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53(6):865-870. doi:10.1136/gut.2003.023937
-
Jones DE, Watt FE, Metcalf JV, Bassendine MF, James OF. Familial primary biliary cirrhosis reassessed: a geographically-based population study. J Hepatol. 1999;30(3):402-407. doi:10.1016/s0168-8278(99)80097-1
-
Gershwin ME, Selmi C, Worman HJ, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42(5):1194-1202. doi:10.1002/hep.20907
-
Floreani A, Franceschet I, Cazzagon N. Primary biliary cirrhosis: overlaps with other autoimmune disorders. Semin Liver Dis. 2014;34(3):352-360. doi:10.1055/s-0034-1383733
-
Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: Convenient and inconvenient truths. Hepatology. 2008;47(2):737-745. doi:10.1002/hep.22042
-
Scheuer PJ. Primary biliary cirrhosis. Proc R Soc Med. 1967;60(12):1257-1260.
-
Poupon RE, Lindor KD, Cauch-Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology. 1997;113(3):884-890. doi:10.1016/s0016-5085(97)70183-5
-
Talwalkar JA, Souto E, Jorgensen RA, Lindor KD. Natural history of pruritus in primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2003;1(4):297-302. doi:10.1016/s1542-3565(03)00134-4
-
Chazouillères O, Wendum D, Serfaty L, Montembault S, Rosmorduc O, Poupon R. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology. 1998;28(2):296-301. doi:10.1002/hep.510280203
-
Berg PA, Klein R. Antimitochondrial antibodies in primary biliary cirrhosis and other disorders: definition and clinical relevance. Dig Dis. 1992;10(2):85-101. doi:10.1159/000171338
-
Corpechot C, Abenavoli L, Rabahi N, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48(3):871-877. doi:10.1002/hep.22428
-
Nevens F, Andreone P, Mazzella G, et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N Engl J Med. 2016;375(7):631-643. doi:10.1056/NEJMoa1509840
-
Corpechot C, Chazouillères O, Rousseau A, et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N Engl J Med. 2018;378(23):2171-2181. doi:10.1056/NEJMoa1714519
Last Updated: 2026-01-05
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Liver Anatomy and Physiology
Differentials
Competing diagnoses and look-alikes to compare.
- Primary Sclerosing Cholangitis
- Autoimmune Hepatitis
- Drug-Induced Liver Injury