Thrombotic Thrombocytopenic Purpura (Adult)
Thrombotic thrombocytopenic purpura (TTP) is a rare, life-threatening thrombotic microangiopathy (TMA) characterised by ... MRCP exam preparation.
What matters first
Thrombotic thrombocytopenic purpura (TTP) is a rare, life-threatening thrombotic microangiopathy (TMA) characterised by ... MRCP exam preparation.
Severe thrombocytopenia (less than 30 x10⁹/L) with schistocytes
8 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Severe thrombocytopenia (less than 30 x10⁹/L) with schistocytes
- Fluctuating neurological symptoms (confusion, seizures, stroke)
- Cardiac ischaemia with elevated troponin
- Refractory to plasma exchange or PEX-dependent
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- Haemolytic Uraemic Syndrome
- Disseminated Intravascular Coagulation
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Thrombotic Thrombocytopenic Purpura (Adult)
1. Overview
Definition and Clinical Significance
Thrombotic thrombocytopenic purpura (TTP) is a rare, life-threatening thrombotic microangiopathy (TMA) characterised by severe deficiency of ADAMTS13 (A Disintegrin and Metalloproteinase with a ThromboSpondin type 1 motif, member 13), the von Willebrand factor (VWF)-cleaving protease. ADAMTS13 activity below 10% is the diagnostic hallmark of TTP. [1]
The condition leads to accumulation of ultra-large VWF multimers in the circulation, causing spontaneous platelet aggregation and widespread microvascular thrombosis. This results in the characteristic triad of microangiopathic haemolytic anaemia (MAHA), severe thrombocytopenia, and organ ischaemia—particularly affecting the brain and heart. [2]
TTP represents a true haematological emergency. Without treatment, mortality exceeds 90%. However, with prompt plasma exchange (PEX), survival rates reach 80-90%, making early recognition and immediate treatment initiation critical. [3] The advent of caplacizumab—an anti-VWF nanobody—and upfront rituximab has transformed outcomes, reducing both acute mortality and long-term relapse rates. [4,5]
Historical Context
TTP was first described by Eli Moschcowitz in 1924 in a 16-year-old girl who died within two weeks of illness onset. The classic "pentad" of thrombocytopenia, MAHA, neurological symptoms, renal impairment, and fever was historically required for diagnosis. However, this complete pentad occurs in only 5-10% of cases, and modern diagnostic criteria focus on the dyad of thrombocytopenia and MAHA in the presence of severe ADAMTS13 deficiency. [6]
The identification of ADAMTS13 in 1996 and its deficiency as the cause of TTP in 2001 revolutionised our understanding and led to targeted therapies. [7]
Why This Matters Clinically
TTP is a high-stakes diagnosis where hours matter. Every day of delay in initiating plasma exchange increases mortality by approximately 1.5-fold. [8] The condition presents in diverse ways—from subtle confusion to fulminant multi-organ failure—and can mimic many other conditions, from stroke to sepsis.
For postgraduate examinations (MRCP, FRACP), TTP is a core topic due to its:
- Diagnostic complexity requiring clinical reasoning
- Emergency management protocols
- Evidence-based treatment advances (HERCULES and TITAN trials)
- Differential diagnosis from other TMAs (aHUS, DIC, HELLP)
- Long-term monitoring and relapse prevention strategies
2. Epidemiology
Incidence and Prevalence
| Statistic | Value | Population | Source |
|---|---|---|---|
| Overall incidence | 1.5-6 per million per year | General population | [9] |
| US incidence | 3.3 per million per year | United States | [10] |
| Peak incidence (African ancestry) | 6.2-12.9 per million per year | African Americans | [10] |
| Acquired (immune) TTP | > 95% of adult cases | Adults | [11] |
| Congenital TTP (USS) | less than 5% of adult cases | Adults | [11] |
The incidence of TTP has increased over recent decades, likely reflecting improved recognition rather than true disease increase. Registry data suggest stable incidence when diagnostic criteria are standardised. [9]
Demographics
Age Distribution
- Peak incidence: 30-50 years (median age ~40 years)
- Can occur at any age from infancy to elderly
- Congenital TTP (Upshaw-Schulman syndrome) often presents in neonatal period or early childhood but may be diagnosed in adulthood
Sex Distribution
- Female predominance: 2-3:1 female-to-male ratio [2]
- Hormonal factors may play a role
- Pregnancy and postpartum period are recognised triggers
Ethnicity
- African ancestry: 3-9 times higher incidence compared to European ancestry [10]
- Possible genetic predisposition (ADAMTS13 gene polymorphisms)
- Hispanic populations also show increased incidence
- Asian populations have lower reported incidence
Geographic Variation
- Higher incidence in United States and Western Europe (likely reflects better ascertainment)
- Registry data available from US (Oklahoma TTP-HUS Registry), UK, France, Italy
Risk Factors and Triggers
Acquired immune TTP is triggered by various factors that precipitate antibody formation against ADAMTS13:
| Trigger/Association | Mechanism | Frequency |
|---|---|---|
| Idiopathic | No identifiable trigger | 40-60% |
| Infection | Immune activation (viral, bacterial) | 20-30% |
| Pregnancy/Postpartum | Immune dysregulation, stress | 10-25% |
| Autoimmune disease (SLE, Sjögren's) | Associated autoantibodies | 10-15% |
| HIV infection | Immune dysregulation | 5-10% |
| Drugs (ticlopidine, quinine, clopidogrel, interferon) | Drug-dependent antibodies | less than 5% |
| Malignancy | Paraneoplastic, immune dysregulation | less than 5% |
| Bone marrow/stem cell transplant | Secondary TMA (often ADAMTS13 > 10%) | Variable |
| Surgery/trauma | Stress, endothelial activation | Rare |
Note: Drug-induced TMA (e.g., ticlopidine 1:1600 risk) often has ADAMTS13 > 10% and represents a distinct entity from immune TTP. [12]
Congenital TTP (Upshaw-Schulman Syndrome) Triggers:
- Infection
- Pregnancy (unmasking in previously undiagnosed patients)
- Surgery
- Physiological stress
3. Aetiology and Pathophysiology
Normal ADAMTS13 Function
ADAMTS13 is a plasma metalloprotease primarily synthesised by hepatic stellate cells and vascular endothelium. [7] Its sole known physiological substrate is ultra-large von Willebrand factor (ULVWF) multimers.
Physiological Role:
- VWF is released from endothelial cells as ULVWF multimers
- Under high shear stress, ULVWF multimers unfold and expose the A2 domain
- ADAMTS13 cleaves the Tyr1605-Met1606 bond in the VWF A2 domain
- This reduces VWF multimer size, preventing excessive platelet adhesion
- Normal ADAMTS13 activity: 50-150%
Classification of TTP
| Type | Mechanism | ADAMTS13 Activity | Inheritance | Frequency (Adults) |
|---|---|---|---|---|
| Acquired Immune TTP | IgG autoantibodies against ADAMTS13 | less than 10% | N/A (acquired) | > 95% |
| Congenital TTP (USS) | Biallelic ADAMTS13 gene mutations | less than 10% (often less than 1%) | Autosomal recessive | less than 5% |
| Secondary TMA | Various (drugs, transplant, pregnancy complications) | Usually > 10% | N/A | Variable |
Pathophysiology of Immune TTP
The pathophysiology follows a cascade from ADAMTS13 deficiency to multi-organ microvascular thrombosis:
Step 1: ADAMTS13 Deficiency
Acquired Immune TTP:
- Polyclonal IgG autoantibodies (rarely IgA, IgM) against ADAMTS13
- Antibodies may:
- Inhibit enzymatic activity (bind to active site or exosite regions)
- Enhance clearance (immune complex formation)
- Both mechanisms often coexist
- Inhibitor titre correlates with disease severity [11]
- CD4+ T-cell response against ADAMTS13 epitopes drives antibody production
Congenital TTP:
- Biallelic mutations in ADAMTS13 gene (chromosome 9q34)
-
200 mutations described (missense, nonsense, frameshift, splice-site)
- Severe deficiency (less than 1% activity) from birth
- Clinical manifestations often triggered by physiological stress
Step 2: Accumulation of Ultra-Large VWF Multimers
- Without ADAMTS13 cleavage, ULVWF multimers persist in circulation
- ULVWF multimers are hyperadhesive for platelets
- Multimer strings can span entire capillary diameter
- Particularly prominent under high shear stress conditions (microvasculature)
Step 3: Spontaneous Platelet Aggregation
- ULVWF multimers bind GPIb-IX-V complex on platelets
- Platelet adhesion and activation occur spontaneously without vessel injury
- Platelet-rich microthrombi form in arterioles and capillaries
- Widespread microvascular occlusion
- Consumption of platelets → severe thrombocytopenia (often less than 30 x10⁹/L)
Key Distinction from DIC:
- Platelets consumed in TTP via VWF-mediated aggregation (not thrombin generation)
- Coagulation cascade NOT activated
- PT, APTT, fibrinogen remain normal (differentiates from DIC)
Step 4: Microangiopathic Haemolytic Anaemia
- Red blood cells (RBCs) encounter platelet-VWF microthrombi in capillaries
- Mechanical shearing of RBCs as they pass through occluded vessels
- RBC fragmentation → schistocytes (helmet cells, fragmented RBCs)
- Intravascular haemolysis:
- ↓ Haemoglobin (typically 60-90 g/L)
- ↑ LDH (often > 1000 U/L, can reach > 2000 U/L)
- ↑ Unconjugated bilirubin
- ↓ Haptoglobin (consumed binding free haemoglobin, often undetectable)
- ↑ Reticulocytes (compensatory bone marrow response)
- Negative direct antiglobulin test (Coombs) distinguishes from AIHA
Step 5: End-Organ Ischaemia
Microthrombi cause organ ischaemia with characteristic patterns:
Brain (Most Characteristic):
- Neurological symptoms in 40-70% [2]
- Fluctuating symptoms (wax and wane with thrombi formation/resolution)
- Manifestations: confusion (40%), headache (55%), seizures (20%), focal deficits, stroke (10%), coma (5%) [13]
- Posterior reversible encephalopathy syndrome (PRES) in 15% [13]
- MRI findings: cortical/subcortical hyperintensities, microhaemorrhages
- Most neurological symptoms reversible with treatment
Heart:
- Cardiac involvement in 30-50% [14]
- Myocardial ischaemia/microinfarction
- Elevated troponin (> 0.1 ng/mL associated with 3-fold mortality increase) [14]
- ECG changes in 40%: ST-segment changes, arrhythmias
- Sudden cardiac death risk 2-5%
- Cardiac biomarkers important prognostic markers
Kidneys:
- Renal involvement typically mild (distinguishes from HUS)
- Elevated creatinine in 20-50%, usually less than 177 μmol/L (2 mg/dL)
- Microhaematuria, proteinuria common
- Severe AKI requiring dialysis suggests alternative diagnosis (aHUS, STEC-HUS)
Other Organs:
- Gastrointestinal: abdominal pain (30%), nausea, pancreatitis
- Pulmonary: rare, but can cause pulmonary hypertension
- Retinal: visual changes, retinal haemorrhages
- Adrenal: rare adrenal insufficiency
Molecular Mechanisms (ExamDetail)
ADAMTS13 Structure and Function:
- 1427 amino acid multidomain protease
- Domains: metalloprotease, disintegrin-like, TSP1 repeats (×8), CUB domains (×2), spacer
- Metalloprotease domain contains catalytic site (requires Zn²⁺, Ca²⁺)
- Spacer domain is major epitope for autoantibodies in acquired TTP
- CUB domains mediate binding to endothelial cells
VWF A2 Domain Cleavage:
- VWF A2 domain normally globular (protected from cleavage)
- High shear stress (> 5000 s⁻¹) unfolds VWF, exposing Tyr1605-Met1606 scissile bond
- ADAMTS13 binds unfolded A2 domain via exosites
- Proteolytic cleavage reduces multimer size from > 10,000 kDa to less than 1,500 kDa
- Deficiency allows ULVWF multimers > 20,000 kDa to persist
Autoantibody Mechanisms:
- Most antibodies target spacer domain (prevent substrate binding)
- Some target CUB domains (enhance clearance)
- Inhibitory antibodies show non-competitive or uncompetitive kinetics
- Antibody-ADAMTS13 complexes cleared by reticuloendothelial system
- T-cell epitopes in spacer and CUB2 domains identified
4. Clinical Presentation
Cardinal Features
The classic pentad (thrombocytopenia, MAHA, neurological symptoms, fever, renal impairment) is present in only 5-10% of cases. [6] Modern diagnosis relies on dyad of thrombocytopenia + MAHA with severe ADAMTS13 deficiency.
Symptom Profile
| Symptom | Frequency | Clinical Characteristics |
|---|---|---|
| Fatigue, weakness | 70-90% | From anaemia; often presenting complaint |
| Neurological symptoms | 40-70% | Fluctuating; wax and wane; includes: |
| - Confusion/altered mental status | 40-50% | Often subtle initially; may progress to coma |
| - Headache | 50-60% | Severe, persistent, or new onset |
| - Seizures | 15-25% | Focal or generalized; may be first presentation |
| - Focal neurological deficits | 10-30% | Hemiparesis, aphasia, visual field defects |
| - Stroke | 5-15% | Ischaemic (thrombotic); haemorrhagic rare |
| - Coma | 5-10% | Poor prognostic sign |
| Bleeding manifestations | 30-60% | Paradoxical (thrombocytopenia despite thrombosis) |
| - Purpura, petechiae | 40-60% | Spontaneous or minimal trauma |
| - Mucosal bleeding | 20-30% | Gingival, epistaxis |
| - Serious bleeding (ICH, GI) | less than 5% | Rare; despite severe thrombocytopenia |
| Gastrointestinal symptoms | 30-50% | Non-specific |
| - Abdominal pain | 30-40% | Mesenteric ischaemia, pancreatitis |
| - Nausea, vomiting | 30-40% | May mimic gastroenteritis |
| - Diarrhoea | 10-20% | If prominent, consider STEC-HUS |
| Chest pain | 15-30% | Cardiac ischaemia; troponin often elevated [14] |
| Fever | 15-30% | Usually low-grade; high fever suggests infection |
| Visual symptoms | 10-20% | Blurred vision, scotomata, diplopia |
Key Clinical Pattern: Symptoms often fluctuate over hours to days, reflecting dynamic thrombi formation and resolution. This fluctuation is highly characteristic of TTP.
Signs on Examination
| System | Findings | Notes |
|---|---|---|
| General appearance | Acute distress, pallor, jaundice | Jaundice often mild (unconjugated hyperbilirubinaemia) |
| Skin | Petechiae, purpura, ecchymoses | Distribution: trunk, extremities, mucous membranes |
| Neurological | Altered consciousness, confusion, focal deficits | GCS may be reduced; fluctuating level |
| Seizure activity, coma (severe) | ||
| Cardiovascular | Tachycardia, flow murmur (anaemia) | Signs of cardiac ischaemia rare but critical |
| Arrhythmias (if cardiac involvement) | ||
| Respiratory | Tachypnoea (compensating metabolic acidosis) | Pulmonary involvement rare |
| Abdominal | Tenderness (non-specific) | Hepatosplenomegaly unusual |
| Fundoscopy | Retinal haemorrhages, exudates | May see papilloedema if severe neurological involvement |
Presentation Patterns by Setting
Emergency Department:
- Confusion + anaemia + thrombocytopenia ("?sepsis, ?stroke")
- Bleeding with low platelets ("?ITP, ?leukaemia")
- Seizures with no prior history
General Medicine Ward:
- Unexplained thrombocytopenia during admission for other reasons
- Falling platelets despite stopping heparin
- Haemolytic anaemia investigation
Haematology Referral:
- Abnormal blood film (schistocytes reported)
- Thrombocytopenia + haemolysis ("MAHA query cause")
Obstetrics:
- Second/third trimester or postpartum TMA [15]
- Differential: HELLP syndrome, preeclampsia, catastrophic APS
Clinical Subtypes
First Episode TTP:
- Idiopathic (no identifiable trigger) in 40-60%
- Acute onset over days to 1-2 weeks
- May have prodromal infection (URI, gastroenteritis)
Relapsing TTP:
- Occurs in 30-60% over 10 years [16]
- May occur months to years after initial episode
- Often preceded by falling ADAMTS13 activity in remission
- Similar clinical features but recognition often faster
Pregnancy-Associated TTP:
- Second/third trimester or within 6 weeks postpartum [15]
- Can be first presentation (acquired) or unmasking of congenital TTP
- Maternal mortality 15-30% if untreated; less than 10% with treatment
- Fetal loss ~30%
- Urgent delivery NOT required (unlike HELLP/preeclampsia if severe)
Congenital TTP (Acute Exacerbation):
- Triggers: infection, pregnancy, surgery, stress
- May have milder presentations if on prophylactic FFP
- Chronic complications: stroke, cognitive impairment from recurrent episodes
5. Differential Diagnosis
TTP is one of several causes of thrombotic microangiopathy (TMA). Distinguishing TTP from other TMAs is critical as treatments differ:
Core Differentials (TMAs)
| Condition | Key Distinguishing Features | ADAMTS13 | Coagulation | Dominant Organ | Treatment |
|---|---|---|---|---|---|
| TTP | Neurological dominant; mild renal; schistocytes; fever rare | less than 10% | Normal PT/APTT | Brain, heart | PEX + caplacizumab + rituximab |
| Atypical HUS (aHUS) | Severe AKI/renal failure; less neuro; genetic testing | > 10% (normal) | Normal PT/APTT | Kidney (AKI, dialysis) | Eculizumab (C5 inhibitor) |
| STEC-HUS (typical HUS) | Diarrhoea prodrome (often bloody); Shiga toxin +; children | > 10% | Normal PT/APTT | Kidney | Supportive; NO antibiotics |
| DIC | Coagulopathy (↑PT/APTT, ↓fibrinogen); underlying trigger (sepsis, malignancy) | > 10% | ↑PT/APTT, ↓fibrinogen | Multi-organ; bleeding | Treat underlying cause |
| HELLP Syndrome | Pregnancy (> 20 weeks); ↑LFTs (AST/ALT); hypertension; ↓platelets | > 10% | Normal or ↑PT/APTT | Liver | Delivery |
| Malignant Hypertension | Severe BP (> 180/120 mmHg); papilloedema; retinopathy | > 10% | Normal PT/APTT | Kidney, brain, heart | Controlled BP reduction |
| Catastrophic APS | Multi-organ thrombosis; arterial/venous; antiphospholipid antibodies + | > 10% | ↑APTT (LA); normal PT | Multi-organ | Anticoagulation + steroids + PEX |
| Malignancy-Associated TMA | Known malignancy (gastric, breast, prostate); bone marrow infiltration | Variable | Normal PT/APTT | Variable | Chemotherapy; PEX limited role |
| Drug-Induced TMA | Exposure to culprit drug (quinine, ticlopidine, gemcitabine, tacrolimus) | Usually > 10% | Normal PT/APTT | Variable | Stop drug; supportive; ±PEX |
Clinical Reasoning Approach [17]:
- Confirm TMA: Thrombocytopenia + MAHA (schistocytes + ↑LDH + ↓haptoglobin) + normal coagulation
- ADAMTS13: less than 10% → TTP; > 10% → other TMA
- Clinical phenotype:
- Neurological dominant → TTP
- Renal failure dominant → HUS
- Diarrhoea prodrome → STEC-HUS
- Pregnancy → HELLP, preeclampsia, TTP, aHUS
- Severe hypertension → malignant HTN
- Coagulopathy → DIC
Other Differentials (Not TMA)
| Condition | Distinguishing Features |
|---|---|
| Immune Thrombocytopenic Purpura (ITP) | Isolated thrombocytopenia; NO haemolysis; NO schistocytes; normal LDH/haptoglobin |
| Autoimmune Haemolytic Anaemia (AIHA) | Haemolysis with positive Coombs; NO schistocytes; normal platelets |
| Evans Syndrome | AIHA + ITP; positive Coombs; NO schistocytes; associated with SLE, lymphoma |
| Sepsis with DIC | Fever, hypotension, source of infection; coagulopathy; normal ADAMTS13 |
| Acute Leukaemia | Blasts on blood film; bone marrow infiltration; pancytopenia |
6. Investigations
Investigations serve to: (1) confirm TMA, (2) measure ADAMTS13, (3) differentiate from other TMAs, (4) assess organ involvement, (5) guide monitoring.
Diagnostic Approach
Immediate (Stat) Tests (within 1-2 hours):
- Full blood count with blood film review
- LDH, bilirubin, haptoglobin, reticulocyte count
- Coagulation screen (PT, APTT, fibrinogen)
- Renal function (creatinine, eGFR)
- Liver function tests
- Troponin (cardiac biomarker)
- Direct antiglobulin test (Coombs)
- Blood group and save
Urgent (Same Day) Tests:
- ADAMTS13 activity (send BEFORE PEX/transfusion)
- ADAMTS13 inhibitor assay
- Complement studies (C3, C4, if considering aHUS)
- Pregnancy test (if applicable)
- HIV, hepatitis serology
Second-Line (Guided by Initial Results):
- ADAMTS13 gene sequencing (if congenital suspected)
- Stool culture, Shiga toxin PCR (if diarrhoea)
- Antiphospholipid antibodies (lupus anticoagulant, anticardiolipin, anti-β2GP1)
- Autoimmune screen (ANA, ENA, dsDNA)
- Malignancy screen (age-appropriate)
Key Investigations
1. Blood Film (Essential Diagnostic Test)
Findings:
- Schistocytes (fragmented RBCs): helmet cells, triangle cells, microspherocytes
- "Quantification: > 1% significant; TTP typically 2-10% [18]"
- "ICSH recommendation: manual count of 1000 RBCs by trained observer"
- Present in 100% of TTP at diagnosis
- Polychromasia (young RBCs/reticulocytes)
- Thrombocytopenia (reduced platelet count on film)
- Nucleated RBCs (severe haemolysis)
Pitfall: Schistocytes may normalise within days of starting PEX; early blood film critical.
2. Laboratory Panel
| Test | Typical Finding in TTP | Interpretation |
|---|---|---|
| Haemoglobin | 60-90 g/L (6-9 g/dL) | Moderate-severe anaemia from MAHA |
| Platelets | less than 30 x10⁹/L (often 10-20) | Severe thrombocytopenia; less than 10 rare |
| Reticulocytes | Elevated (> 100 x10⁹/L or > 2%) | Compensatory response to haemolysis |
| LDH | Markedly elevated (> 1000 U/L, often > 2000) | Intravascular haemolysis + tissue ischaemia |
| Haptoglobin | Undetectable (less than 0.1 g/L) | Consumed binding free haemoglobin |
| Bilirubin | ↑ Unconjugated (30-60 μmol/L) | Haemolysis; mild jaundice |
| Direct Coombs (DAT) | Negative | Excludes AIHA; key differentiator |
| PT | Normal | Distinguishes from DIC |
| APTT | Normal | Distinguishes from DIC |
| Fibrinogen | Normal or slightly ↑ | Distinguishes from DIC (↓ in DIC) |
| D-Dimer | May be elevated | Non-specific; elevated in thrombosis/haemolysis |
| Creatinine | Normal or mildly ↑ (less than 177 μmol/L) | Mild renal involvement (cf. severe in HUS) |
| Troponin | Elevated in 30-50% | Cardiac ischaemia; poor prognostic marker [14] |
| AST/ALT | Mildly elevated | Ischaemia; marked elevation suggests HELLP |
3. ADAMTS13 Assays (Gold Standard)
ADAMTS13 Activity:
- Diagnostic threshold: less than 10% activity [1]
- Functional assay measures VWF cleavage (FRETS-VWF73 commonly used)
- Turnaround time: 1-5 days (varies by lab)
- Critical: Sample BEFORE plasma exchange or blood transfusion (dilutes result)
- Sample: Citrated plasma (purple/blue top bottle)
| ADAMTS13 Activity | Interpretation | Clinical Action |
|---|---|---|
| less than 10% | TTP (acquired or congenital) | Diagnoses TTP; continue treatment |
| 10-30% | Partial deficiency; not diagnostic of TTP | Consider other TMA; may indicate recovering TTP or increased relapse risk |
| > 30% | Normal or mildly reduced | Excludes TTP; investigate other TMA causes |
ADAMTS13 Inhibitor/Antibody:
- Detects IgG autoantibodies against ADAMTS13
- Positive: Acquired immune TTP (> 95% of adult cases)
- Negative with ADAMTS13 less than 10%: Suggests congenital TTP (require genetic testing)
- Inhibitor titre (Bethesda units): Higher titres correlate with severity [11]
Genetic Testing:
- Indicated if ADAMTS13 less than 10% with negative inhibitor
- Biallelic ADAMTS13 mutations confirm Upshaw-Schulman syndrome
- Implications: lifelong prophylaxis, genetic counselling, pregnancy planning
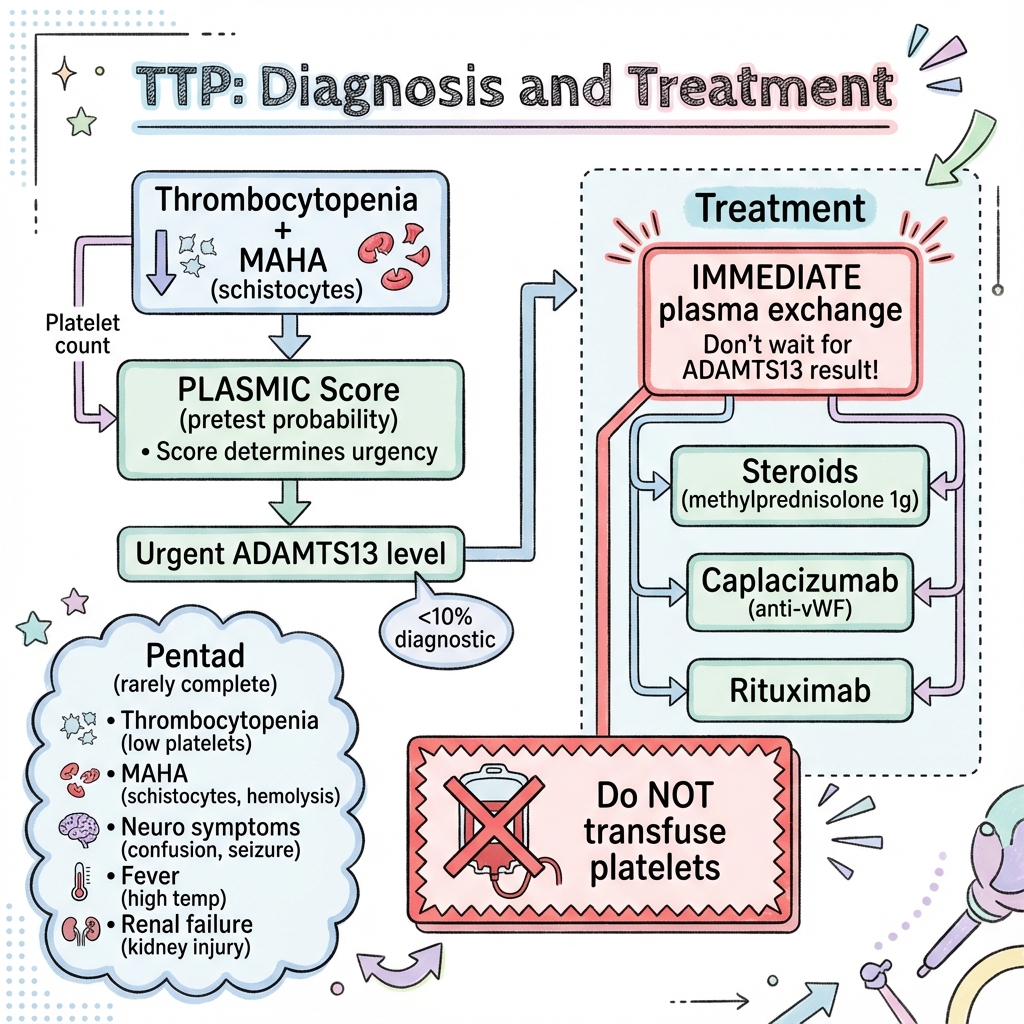
4. PLASMIC Score (Clinical Prediction Tool)
The PLASMIC score estimates pretest probability of severe ADAMTS13 deficiency before assay results are available: [19]
| Variable | Points |
|---|---|
| Platelet count less than 30 x10⁹/L | 1 |
| Combined haemolysis variables (↑reticulocytes OR ↓haptoglobin OR ↑indirect bilirubin) | 1 |
| Absence of active Cancer | 1 |
| Absence of Stem cell or solid organ transplant | 1 |
| MCV less than 90 fL | 1 |
| INR less than 1.5 | 1 |
| Creatinine less than 177 μmol/L (2.0 mg/dL) | 1 |
| Total | 0-7 |
Interpretation [19]:
- Score 6-7 (High risk): 72% have ADAMTS13 less than 10%; sensitivity 90%, specificity 92%
- Score 5 (Intermediate risk): ~40% have ADAMTS13 less than 10%
- Score 0-4 (Low risk): 0-4% have ADAMTS13 less than 10%; TTP very unlikely
Clinical Use: PLASMIC ≥5 warrants immediate empiric treatment while awaiting ADAMTS13 results.
5. Imaging
Brain MRI (if neurological symptoms):
- Indications: confusion, seizures, focal deficits, coma
- Findings:
- Cortical/subcortical T2/FLAIR hyperintensities (reversible)
- Microhaemorrhages (susceptibility-weighted imaging)
- Posterior reversible encephalopathy syndrome (PRES) pattern in 15% [13]
- Ischaemic strokes (10%)
- Microinfarcts
- Most changes reversible with treatment
Cardiac MRI (if troponin elevated):
- May show microinfarcts, oedema
- Prognostic value
CT Head (emergency if altered consciousness):
- Exclude intracranial haemorrhage (before PEX line insertion)
- Less sensitive than MRI for microangiopathic changes
7. Classification and Staging
Classification by Aetiology
| Type | ADAMTS13 | Inhibitor | Age of Onset | Treatment Approach |
|---|---|---|---|---|
| Acquired Immune TTP | less than 10% | Positive | Any age (peak 30-50) | PEX + immunosuppression |
| Congenital TTP (USS) | less than 10% (often less than 1%) | Negative | Neonatal/childhood (may present in adults) | FFP replacement (no immunosuppression) |
Classification by Presentation
- First episode: No prior history of TTP
- Relapse: Recurrent TTP after > 30 days of remission (platelets normalised)
- Exacerbation: Recurrence within 30 days of stopping PEX (before remission achieved)
Severity Assessment
No validated severity score exists, but prognostic markers include:
Poor Prognostic Markers:
- Cardiac involvement (troponin > 0.1 ng/mL): 3-fold ↑ mortality [14]
- Coma (GCS less than 8)
- Age > 60 years
- High inhibitor titre
- Delay to treatment > 24 hours [8]
- PEX-refractory disease
Good Prognostic Markers:
- Early recognition and treatment
- Platelet response within 5 days of PEX
- ADAMTS13 recovery
8. Management
TTP is a medical emergency. Treatment must be initiated immediately based on clinical suspicion (thrombocytopenia + MAHA ± neurological symptoms) without waiting for ADAMTS13 results. Delay increases mortality. [8]
Immediate Emergency Management (Within 2 Hours)
1. Resuscitation (ABCDE Approach):
- Airway: Secure if GCS less than 8 (coma)
- Breathing: Oxygen if hypoxic
- Circulation: IV access (×2 large-bore cannulae); fluid resuscitation if needed
- Disability: GCS assessment; exclude hypoglycaemia
- Exposure: Examine for purpura, petechiae
2. Initiate Emergency Treatments:
| Intervention | Details | Grade |
|---|---|---|
| Plasma Exchange (PEX) | 1-1.5 plasma volumes (40-60 mL/kg) daily via central venous catheter | 1A [20] |
| Corticosteroids | Methylprednisolone 1g IV daily × 3 days, OR Prednisolone 1 mg/kg PO daily | 2B [20] |
| Caplacizumab | 11 mg IV bolus (before first PEX), then 11 mg SC daily | 2B [20] |
3. Urgent Haematology Consultation:
- Contact on-call haematologist immediately
- Discuss PEX access and initiation
- Arrange apheresis team
4. Obtain Investigations (before PEX if possible):
- ADAMTS13 activity and inhibitor (CRITICAL: sample before PEX)
- All other investigations as per Section 6
Plasma Exchange (PEX) — The Life-Saving Intervention
Mechanism of Action [8]:
- Removes: IgG autoantibodies, ultra-large VWF multimers, inflammatory cytokines
- Replaces: ADAMTS13 (from donor plasma), coagulation factors
- Rapidly corrects microvascular thrombosis
Technical Details:
- Volume: 1-1.5 plasma volumes (approximately 40-60 mL/kg; ~3000 mL for 70 kg adult)
- Replacement fluid: Fresh frozen plasma (FFP) or solvent-detergent plasma
- Frequency: Daily until platelet count normalises (> 150 x10⁹/L on 2 consecutive days)
- Access: Central venous catheter (tunnelled or non-tunnelled; internal jugular/femoral preferred)
- Duration: Median 5-9 days (shorter with caplacizumab) [4,5]
- Anticoagulation: Citrate-based (monitor ionised calcium; risk of citrate toxicity)
Evidence:
- Mortality without PEX: > 90%
- Mortality with PEX: 10-20% [3]
- Delay > 24 hours: 1.5-fold increase in mortality [8]
- 30-day survival: 85-95% [8]
Monitoring During PEX:
- Daily platelet count, LDH (markers of response)
- Daily Hb, reticulocytes, bilirubin, haptoglobin
- Ionised calcium (citrate toxicity)
- Fluid balance
- Signs of line infection
Stopping PEX:
- Criteria: Platelets > 150 x10⁹/L on 2 consecutive days + improving LDH
- Do NOT stop prematurely (risk of exacerbation)
- Taper may be considered (e.g., reduce frequency to alternate days before stopping)
Caplacizumab — Anti-VWF Nanobody
Caplacizumab is a bivalent anti-VWF nanobody that prevents VWF-platelet binding, addressing the pathophysiology directly. [4,5]
Dosing:
- Loading dose: 11 mg IV bolus (before first PEX)
- Maintenance: 11 mg SC daily during PEX and for 30 days post-PEX
- Supplied as pre-filled syringes
Evidence:
- HERCULES Trial (2019) [4]: Caplacizumab vs placebo (both with PEX + steroids)
- "Primary endpoint: Time to platelet normalisation: 74% vs 49% by day 5 (pless than 0.001)"
- "Composite endpoint (death/TTP recurrence/thromboembolic event): 12% vs 38% (pless than 0.001)"
- "Number needed to treat: 4 (highly effective)"
- TITAN Trial (2016) [5]: Median platelet normalisation 2.97 vs 4.79 days; fewer exacerbations
Advantages:
- Faster platelet recovery (reduces ICU time, PEX sessions)
- Reduced TTP-related death and recurrence
- Reduced thromboembolic complications
Adverse Effects:
- Bleeding risk: Inhibits VWF function; epistaxis, gingival bleeding (usually minor)
- Major bleeding rare (2-4%); monitor closely if platelets less than 30 x10⁹/L
- Antibody formation (~5%; rarely neutralising)
Licensing: Approved by EMA (2018), FDA (2019) for acquired TTP.
Corticosteroids
Mechanism: Suppress autoantibody production; reduce endothelial activation. [21]
Regimens:
- High-dose IV: Methylprednisolone 1g IV daily × 3 days, then oral taper
- Moderate-dose oral: Prednisolone 1 mg/kg PO daily (~60-80 mg)
Taper:
- After platelet normalisation, taper gradually over 6-8 weeks
- Monitor ADAMTS13 activity during taper
- Avoid abrupt cessation (risk of relapse)
Evidence: No RCT evidence, but standard practice based on observational data; improves ADAMTS13 recovery.
Rituximab — B-Cell Depletion
Rituximab (anti-CD20 monoclonal antibody) targets B-cells producing anti-ADAMTS13 antibodies. [22,23]
Indications:
- Upfront (first-line): ISTH guidelines suggest considering in all patients (Grade 2C) [20]
- Refractory disease: Suboptimal response to PEX by day 5
- Relapse prevention: History of relapse
- Preemptive: ADAMTS13 activity falling in remission (less than 10-20%)
Dosing:
- Standard: 375 mg/m² IV weekly × 4 doses
- Alternative: Single dose 1000 mg or lower doses (efficacy data emerging)
Evidence:
- Meta-analysis (2020) [22]: Reduces relapse from 50% to 10-20%; no increase in infections
- Westwood et al. (2013) [23]: Upfront rituximab reduced relapse at 1 year from 57% to 10% (p=0.001); fewer PEX sessions (median 5 vs 9)
Timing:
- Can start during acute phase (alongside PEX + steroids + caplacizumab)
- Weekly infusions (4 weeks)
Monitoring:
- CD19 count (B-cell depletion)
- Immunoglobulin levels (risk of hypogammaglobulinaemia)
- Infection surveillance
Adverse Effects:
- Infusion reactions (premedicate with paracetamol, antihistamine)
- Infections (respiratory, herpes zoster reactivation)
- Rarely: progressive multifocal leukoencephalopathy (PML; extremely rare in TTP)
Refractory TTP (5-10% of Cases)
Definition [24]:
- Platelet count fails to normalise by day 5-7 despite daily PEX
- Platelet-dependent (requires continued PEX to maintain platelets)
Management Options:
- Twice-daily PEX: Increase to 1.5 plasma volumes BID
- Rituximab: If not already given (375 mg/m² weekly × 4)
- Cyclosporine: 3-5 mg/kg/day PO (target trough 100-200 ng/mL); alternative calcineurin inhibitor
- Bortezomib: 1.3 mg/m² IV (days 1, 4, 8, 11); targets plasma cells producing autoantibodies
- Vincristine: 1.4 mg/m² IV (max 2 mg) weekly
- N-acetylcysteine: 150 mg/kg IV loading, then infusion (reduces VWF multimer size; experimental)
- Splenectomy: Rarely used now (removed source of antibody production; risk of post-splenectomy sepsis)
Prognosis: Refractory disease carries higher mortality; aggressive multimodal therapy required.
Supportive Care
| Intervention | Details |
|---|---|
| Red cell transfusion | Target Hb > 70 g/L (restrictive strategy); use if symptomatic anaemia |
| Platelet transfusion | Avoid unless life-threatening bleeding (ICH, massive GI bleed); historical concern of worsening thrombosis; use lowest dose if essential [25] |
| Folic acid | 5 mg PO daily (haemolysis increases folate turnover) |
| VTE prophylaxis | Avoid until platelets recover > 50 x10⁹/L; then LMWH or mechanical prophylaxis |
| Anticonvulsants | If seizures occur; levetiracetam or lorazepam |
| Antihypertensives | Control BP if hypertensive |
| Avoid antiplatelet agents | Stop aspirin, clopidogrel (bleeding risk) |
| Infection prophylaxis | If on high-dose steroids + rituximab: PCP prophylaxis (co-trimoxazole), antifungal if high risk |
Monitoring Treatment Response
Daily Monitoring (during acute phase):
- Platelet count (aim for normalisation > 150 x10⁹/L)
- LDH (should decline; persistent elevation suggests ongoing haemolysis)
- Haemoglobin, reticulocytes
- Creatinine, electrolytes
- Neurological status
Weekly Monitoring:
- ADAMTS13 activity (should recover to > 30% ideally)
- Inhibitor titre (should decline)
Remission Criteria:
- Platelet count > 150 x10⁹/L on 2 consecutive days
- Normalising LDH
- Resolution of haemolysis
- Clinical improvement (neurological symptoms resolving)
Special Populations
Pregnancy [15]:
- TTP can occur in 2nd/3rd trimester or postpartum
- PEX is safe and effective in pregnancy
- Caplacizumab: limited data; case reports suggest safety
- Delivery NOT urgently required (unlike HELLP/preeclampsia)
- Differentiate from HELLP, preeclampsia, catastrophic APS
- Multidisciplinary care: Haematology + Obstetrics
Congenital TTP (Upshaw-Schulman Syndrome) [26]:
- Acute exacerbation: FFP infusion (10-15 mL/kg every 2-3 weeks) OR plasma exchange
- Prophylaxis: Regular FFP infusions (prevent acute episodes)
- NO role for immunosuppression (steroids, rituximab) — not immune-mediated
- Pregnancy: High-risk; requires increased FFP frequency; close monitoring
- Genetic counselling: Autosomal recessive; partner testing if planning children
9. Long-Term Management and Relapse Prevention
Post-Remission Monitoring
ADAMTS13 Activity Surveillance [27]:
- Frequency:
- Monthly for first 6 months
- Every 3 months for next 6-12 months
- Every 6-12 months thereafter (lifelong)
- Risk stratification:
- "ADAMTS13 less than 20% in remission: 3-fold increased relapse risk [27]"
- "Rising inhibitor titres: 5-fold increased relapse risk [27]"
Clinical Review:
- Monitor for symptoms of relapse (fatigue, bruising, headache, confusion)
- Annual full blood count, LDH, renal function
- Cardiovascular risk assessment (stroke, MI risk)
- Cognitive assessment (neuropsychology if deficits)
Relapse Prevention Strategies
Relapse Epidemiology:
- Overall relapse risk: 30-60% at 10 years [16]
- Median time to relapse: 1-2 years (range: months to decades)
- Most relapses occur within 5 years
Interventions to Reduce Relapse:
-
Upfront Rituximab [22,23]:
- Reduces relapse from 50% to 10-20%
- Longer time to relapse
- Standard in many centres
-
Preemptive Rituximab [27]:
- If ADAMTS13 less than 10-20% during remission (without clinical TTP)
- Prevents progression to acute TTP in 60%
- Single dose (1000 mg) or standard 4-week course
-
Avoid Triggers:
- Infection: Vaccinations (influenza, pneumococcal), infection avoidance
- Smoking cessation
- Manage cardiovascular risk factors
-
Patient Education:
- Recognise early symptoms (fatigue, bruising, confusion)
- Seek immediate medical attention if suspected relapse
- Wear medical alert bracelet/card
Management of Relapse
- Same treatment as first episode: PEX + steroids + caplacizumab + rituximab
- Response often faster (recognition quicker)
- Consider more aggressive immunosuppression if multiple relapses (e.g., prolonged rituximab, cyclosporine)
10. Complications
| Complication | Incidence | Mechanism | Management |
|---|---|---|---|
| Death | 10-20% (with treatment); > 90% (without) [3] | Multi-organ failure, cardiac ischaemia, stroke | Early aggressive PEX + adjuncts |
| Stroke | 5-15% [13] | Ischaemic (microthrombi); rarely haemorrhagic | Acute stroke care; thrombolysis contraindicated (thrombocytopenia); PEX |
| Myocardial infarction | 2-5% | Coronary microthrombosis | Cardiac monitoring; troponin surveillance; PEX; avoid antiplatelet agents initially |
| Sudden cardiac death | 2-5% [14] | Arrhythmias, microinfarcts | Cardiac monitoring; correct electrolytes; PEX |
| Seizures | 15-25% [13] | Cerebral microinfarcts, PRES | Anticonvulsants (levetiracetam); PEX |
| Coma | 5-10% | Severe cerebral involvement | ICU care; airway protection; PEX |
| Cognitive impairment (long-term) | 25-40% [16] | Recurrent microinfarcts | Neuropsychology; cognitive rehabilitation; relapse prevention |
| Depression, fatigue (long-term) | 30-50% [16] | Multifactorial (organic brain injury, psychological trauma) | Psychiatric support; antidepressants; counselling |
| Relapse | 30-60% at 10 years [16] | Recurrent autoantibody formation | ADAMTS13 monitoring; preemptive rituximab; patient education |
| PEX-related complications | 5-15% | Line insertion, catheter infection, citrate toxicity, allergic reactions | Line care; citrate monitoring; premedication for FFP reactions |
| Bleeding (caplacizumab) | 2-4% major; 10-20% minor [4] | VWF inhibition | Monitor closely; dose interruption if major bleeding |
| Infections (rituximab) | 5-10% | B-cell depletion, immunosuppression | Prophylaxis (PCP); vaccination (delayed until B-cell recovery); early antibiotics |
11. Prognosis
Acute Phase Outcomes
| Outcome | Rate | Notes |
|---|---|---|
| Survival (with modern treatment) | 80-90% [3] | PEX + caplacizumab + rituximab |
| 30-day survival | 85-95% [8] | Improved with early PEX |
| Platelet normalisation | Median 5-9 days | Faster with caplacizumab (2-3 days) [4] |
| ICU admission | 30-50% | Coma, cardiac involvement, severe illness |
| Neurological recovery | 70-80% complete | 20-30% persistent deficits [13,16] |
Long-Term Outcomes
| Outcome | Rate | Timeframe |
|---|---|---|
| Relapse | 30-60% | 10 years [16] |
| Relapse (with rituximab) | 10-20% | 5-10 years [22] |
| 10-year survival | 70-85% [16] | Accounting for relapses |
| Cognitive impairment | 25-40% [16] | Persistent; affects QOL |
| Depression | 30% [16] | Long-term psychological impact |
| Chronic fatigue | 50% [16] | Multifactorial |
| Cardiovascular events (stroke, MI) | Increased risk | Long-term; mechanism unclear |
Prognostic Factors
Favourable:
- Early diagnosis and treatment initiation [8]
- Platelet response within 5 days
- ADAMTS13 recovery to > 30%
- Use of caplacizumab [4]
- Upfront rituximab [22,23]
- Younger age
Unfavourable:
- Cardiac involvement (troponin > 0.1 ng/mL): 3-fold mortality [14]
- Coma (GCS less than 8)
- Age > 60 years
- Delayed treatment > 24-48 hours [8]
- Refractory disease (no platelet response by day 5-7)
- High inhibitor titre
- Multiple relapses
12. Prevention and Screening
Primary Prevention
No established primary prevention (unpredictable autoantibody formation).
Risk Reduction:
- Infection prevention (vaccinations: influenza, pneumococcal)
- Cardiovascular risk modification (post-TTP)
Secondary Prevention (Relapse Prevention)
See Section 9: Long-Term Management and Relapse Prevention.
Key Strategies:
- ADAMTS13 monitoring [27]
- Preemptive rituximab if ADAMTS13 less than 10-20%
- Patient education
- Avoid triggers
Screening
ADAMTS13 Screening (not routine population screening):
- Family members of congenital TTP patients (if considering having children)
- Recurrent pregnancy loss with TMA features
- Unexplained neonatal jaundice/thrombocytopenia
13. Guidelines and Evidence Base
Key Guidelines
-
International Society on Thrombosis and Haemostasis (ISTH) Guidelines (2020) [20]
- Zheng XL, et al. J Thromb Haemost. 2020;18(10):2496-2502. PMID: 32239796
- Grade 1A: Immediate PEX for suspected TTP
- Grade 2B: Corticosteroids as adjunct
- Grade 2C: Consider upfront rituximab
- Grade 2B: Caplacizumab in severe cases
-
British Society for Haematology (BSH) Guidelines (2012, updated 2023) [3]
- Scully M, et al. Br J Haematol. 2012;158(3):323-335. PMID: 22624596
- Emphasises immediate PEX without delay
- Avoid platelet transfusion unless life-threatening bleeding
- 1.5 plasma volumes daily; continue until platelets > 150 x2 days
-
American Society of Apheresis (ASFA) Guidelines
- PEX for TTP: Category I (first-line, standard of care)
Landmark Trials and Studies
| Study | Year | Key Findings | PMID |
|---|---|---|---|
| HERCULES Trial [4] | 2019 | Caplacizumab: 74% vs 49% platelet normalisation less than 5 days; 12% vs 38% composite endpoint (death/recurrence/TE); NNT=4 | 30625035 |
| TITAN Trial [5] | 2016 | Caplacizumab: faster platelet recovery (2.97 vs 4.79 days); fewer exacerbations | 26833331 |
| Westwood et al. (Rituximab) [23] | 2013 | Upfront rituximab: 10% vs 57% relapse at 1 year; fewer PEX sessions | 25398525 |
| Bendapudi et al. (PLASMIC Score) [19] | 2017 | PLASMIC ≥5: 90% sensitivity, 92% specificity for ADAMTS13 less than 10%; validated prediction tool | 28259520 |
| Hie et al. (Rituximab Meta-analysis) [22] | 2020 | Rituximab reduces relapse from 50% to 10-20%; no increased infections | 32909630 |
| Benhamou et al. (Cardiac Involvement) [14] | 2019 | Troponin > 0.1: 3-fold mortality; cardiac involvement 30-50% | 31570614 |
| Han et al. (Long-term Outcomes) [16] | 2021 | 25-40% persistent cognitive impairment; 30-60% relapse at 10 years; QOL reduced | 33508122 |
| Zheng et al. (Pathophysiology Review) [1] | 2023 | Comprehensive iTTP review: mortality 90%→10% with PEX; ADAMTS13 less than 10% diagnostic | 36752736 |
Key Evidence Synthesis
Evolution of TTP Mortality:
- Pre-1980 (no PEX): > 90% mortality
- 1980s-2000s (PEX era): 10-20% mortality [3]
- 2016-present (caplacizumab era): less than 10% mortality, faster recovery [4,5]
- 2010s-present (rituximab era): Relapse reduced from 50% to 10-20% [22,23]
Paradigm Shifts:
- 1924: Moschcowitz describes pentad
- 1982: PEX introduced; transforms survival
- 2001: ADAMTS13 identified as causative deficiency
- 2012: Rituximab enters standard practice
- 2016-2019: Caplacizumab trials (TITAN, HERCULES) establish benefit
- 2020: ISTH guidelines codify modern multimodal therapy
14. Differential Diagnosis: TTP vs HUS vs DIC (Summary Table)
| Feature | TTP | Atypical HUS | STEC-HUS | DIC |
|---|---|---|---|---|
| ADAMTS13 | less than 10% | > 10% (normal) | > 10% (normal) | > 10% (normal) |
| Neurological | +++++ (dominant) | + (mild) | + (mild) | + (encephalopathy) |
| Renal | + (mild AKI) | +++++ (severe AKI, dialysis) | +++++ (severe AKI) | ++ (variable) |
| Coagulation | Normal PT/APTT | Normal PT/APTT | Normal PT/APTT | ↑PT/APTT, ↓fibrinogen |
| Age | Adults (30-50) | Children + adults | Children less than 5 years | Any (underlying trigger) |
| Prodrome | Infection (variable) | None/infection | Diarrhoea (bloody) | Sepsis, malignancy, trauma |
| Treatment | PEX + caplacizumab + rituximab | Eculizumab (C5 inhibitor) | Supportive (NO antibiotics) | Treat underlying cause |
| Pathophysiology | ADAMTS13 deficiency → ULVWF → platelet thrombi | Complement dysregulation | Shiga toxin → endothelial damage | Thrombin generation → DIC |
15. Patient Information and Communication
Explaining TTP to Patients
Simple Explanation: "You have a condition called TTP where your blood is forming tiny clots in small blood vessels throughout your body. This is happening because your body is attacking a protein that normally prevents clotting. The clots use up your platelets (cells that stop bleeding) and damage your red blood cells. This can affect your brain, heart, and other organs."
Treatment Explanation: "The main treatment is called plasma exchange. We filter your blood through a machine, removing the harmful antibodies and replacing your plasma (the liquid part of blood). This, combined with medications, stops the clotting and allows your body to recover. Treatment usually takes 1-2 weeks in hospital."
Prognosis Discussion: "With treatment, most people (80-90%) recover well. However, TTP can come back in about 30-50% of people over the next 10 years, so we'll monitor you closely with regular blood tests. We'll check your ADAMTS13 levels every few months to catch any early signs of relapse."
Warning Signs for Patients (Post-Discharge)
Seek immediate medical attention if you develop:
- New or worsening confusion, headache, or visual changes
- Seizures
- Chest pain or shortness of breath
- Unexplained bruising or bleeding
- Severe fatigue or weakness
- Dark urine or jaundice
Advise:
- Carry medical alert card/bracelet stating "TTP history"
- Inform all healthcare providers of TTP history
- Attend all follow-up appointments
- Comply with ADAMTS13 monitoring
16. Examination Focus (Viva and Written Exams)
Opening Statement (Viva)
Examiner: "Tell me about thrombotic thrombocytopenic purpura."
Model Answer: "TTP is a life-threatening thrombotic microangiopathy caused by severe ADAMTS13 deficiency, defined as activity less than 10%. ADAMTS13 is the VWF-cleaving protease; its deficiency leads to accumulation of ultra-large VWF multimers, causing spontaneous platelet aggregation and microvascular thrombosis. This results in severe thrombocytopenia, microangiopathic haemolytic anaemia with schistocytes, and organ ischaemia—particularly neurological and cardiac. Most adult cases are acquired immune TTP from IgG autoantibodies. Without treatment, mortality exceeds 90%, but with emergency plasma exchange, survival is 80-90%. Modern treatment includes PEX, corticosteroids, caplacizumab, and rituximab, based on ISTH 2020 guidelines and the HERCULES trial."
High-Yield Exam Facts
Diagnostic:
- ADAMTS13 less than 10% = TTP [1]
- PLASMIC score ≥5: high probability TTP [19]
- Schistocytes > 1% on blood film (100% sensitive) [18]
- Normal PT/APTT/fibrinogen (distinguishes from DIC)
- Negative Coombs (distinguishes from AIHA)
Clinical:
- Complete pentad only 5-10% [6]
- Dyad (thrombocytopenia + MAHA) sufficient to start treatment
- Neurological symptoms 40-70% (hallmark of TTP vs other TMAs) [2]
- Fever only 15-30% (not common) [6]
Treatment:
- Emergency PEX within 2 hours (Grade 1A) [20]
- Never delay PEX for ADAMTS13 result
- Avoid platelet transfusion (unless life-threatening bleeding) [25]
- Caplacizumab: NNT=4 (HERCULES trial) [4]
- Rituximab reduces relapse 50% → 10-20% [22]
Outcomes:
- Troponin > 0.1: 3-fold mortality [14]
- Delay > 24 h: 1.5-fold mortality [8]
- Relapse 30-60% at 10 years [16]
Common Exam Questions
-
"How do you differentiate TTP from HUS?"
- ADAMTS13 (less than 10% in TTP, normal in HUS)
- Neurological dominant (TTP) vs renal dominant (HUS)
- Adults (TTP) vs children (STEC-HUS)
- Severe AKI/dialysis suggests HUS, not TTP
-
"Why do you not give platelets in TTP?"
- Historical teaching: platelets "add fuel to fire" and worsen microthrombosis
- Retrospective data suggest possible increased mortality/neurological events [25]
- Current guidelines: avoid unless life-threatening bleeding (ICH, massive GI bleed)
- If given, use lowest dose necessary
-
"What is the PLASMIC score?"
- Clinical prediction tool for ADAMTS13 less than 10% before lab results available [19]
- 7 variables (platelets less than 30, haemolysis, no cancer, no transplant, MCV less than 90, INR less than 1.5, Cr less than 177)
- Score ≥5: 72% have ADAMTS13 less than 10%; sensitivity 90%, specificity 92%
- Use to justify empiric PEX while awaiting ADAMTS13
-
"Describe the evidence for caplacizumab."
- HERCULES trial (2019): 145 patients, RCT, caplacizumab vs placebo [4]
- 74% vs 49% platelet normalisation less than 5 days (pless than 0.001)
- 12% vs 38% composite endpoint (death/recurrence/TE) (pless than 0.001)
- NNT = 4; licenced EMA 2018, FDA 2019
-
"What is your approach to a patient with thrombocytopenia, anaemia, and schistocytes?"
- Confirm TMA: blood film (schistocytes), LDH (high), haptoglobin (low), Coombs (negative), coagulation (normal)
- Calculate PLASMIC score
- Send ADAMTS13 activity and inhibitor (before PEX)
- If PLASMIC ≥5 or high clinical suspicion: start PEX immediately (do not wait for ADAMTS13)
- Add steroids, caplacizumab, rituximab
- Avoid platelet transfusion
- Daily monitoring: platelets, LDH
- Continue PEX until platelets > 150 x2 days
Common Mistakes (What Fails Candidates)
❌ Waiting for ADAMTS13 result before starting PEX → Delays treatment; increases mortality ❌ Transfusing platelets routinely → Worsens thrombosis (avoid unless life-threatening bleeding) ❌ Diagnosing TTP with ADAMTS13 > 10% → Not TTP; investigate other TMA ❌ Confusing TTP with HUS → Different treatments (PEX vs eculizumab) ❌ Missing cardiac involvement → Troponin is critical prognostic marker [14] ❌ Forgetting to send ADAMTS13 before PEX → Dilutes sample; false-normal result ❌ Stopping PEX too early → Risk of exacerbation; need platelets > 150 x2 days
Viva Anchors (Key Phrases to Use)
- "ADAMTS13 less than 10% is diagnostic"
- "Emergency plasma exchange is life-saving and should not be delayed"
- "PLASMIC score guides empiric treatment"
- "Neurological features distinguish TTP from HUS"
- "Normal coagulation distinguishes TTP from DIC"
- "Caplacizumab has a number needed to treat of 4 based on HERCULES trial"
- "Rituximab reduces relapse from 50% to 10-20%"
- "Troponin elevation predicts 3-fold increased mortality"
- "Avoid platelet transfusion unless life-threatening bleeding"
17. References
-
Zheng XL, et al. Immune thrombotic thrombocytopenic purpura: pathophysiology, diagnosis, and management. J Clin Invest. 2023;133(3):e165900. PMID: 36752736
-
Joly BS, et al. Thrombotic thrombocytopenic purpura: a contemporary review. Blood Rev. 2022;54:100927. PMID: 35316270
-
Scully M, et al. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol. 2012;158(3):323-335. PMID: 22624596
-
Scully M, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380(4):335-346. PMID: 30625035
-
Peyvandi F, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2016;374(6):511-522. PMID: 26833331
-
Joly BS, et al. Clinical presentation of TTP: the changing pentad. Semin Hematol. 2014;51(3):236-242. PMID: 25264959
-
Lenting PJ, et al. ADAMTS13 biology and disease. J Thromb Haemost. 2021;19(2):348-359. PMID: 33909823
-
Azoulay E, et al. Therapeutic plasma exchange in critically ill patients with thrombotic thrombocytopenic purpura. Intensive Care Med. 2019;45(8):1153-1155. PMID: 31292116
-
Joly BS, et al. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-2846. PMID: 28416507
-
Deford CC, et al. Multiple major morbidities and increased mortality during long-term follow-up after recovery from thrombotic thrombocytopenic purpura. Blood. 2013;122(12):2023-2029. PMID: 23838348
-
Lenting PJ, et al. An emerging view on inhibitory antibodies in hemophilia A: it takes two to tango. J Thromb Haemost. 2015;13(Suppl 1):S44-52. PMID: 26149048
-
Al-Nouri ZL, et al. Drug-induced thrombotic microangiopathy: a systematic review of published reports. Blood. 2015;125(4):616-618. PMID: 25414441
-
Miller JB, et al. Neurological manifestations of thrombotic thrombocytopenic purpura. J Neurol Sci. 2020;414:116843. PMID: 32405340
-
Benhamou Y, et al. Cardiac involvement is frequent in patients with thrombotic thrombocytopenic purpura and is associated with an adverse outcome. Circulation. 2019;140(13):1065-1067. PMID: 31570614
-
Moatti-Cohen M, et al. Unexpected frequency of Upshaw-Schulman syndrome in pregnancy-onset thrombotic thrombocytopenic purpura. Blood. 2012;119(24):5888-5897. PMID: 22547583
-
Han B, et al. Long-term outcomes and quality of life of patients with thrombotic thrombocytopenic purpura. Blood Adv. 2021;5(3):781-790. PMID: 33508122
-
George JN, et al. Distinguishing thrombotic thrombocytopenic purpura from other microangiopathies. Blood Rev. 2021;48:100792. PMID: 34265830
-
Lesesve JF, et al. Schistocytes in disseminated intravascular coagulation. Int J Lab Hematol. 2014;36(4):439-443. PMID: 24750675
-
Bendapudi PK, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4(4):e157-e164. PMID: 28259520
-
Zheng XL, et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2496-2502. PMID: 32239796
-
Li J, et al. The role of corticosteroids in thrombotic thrombocytopenic purpura. Ther Apher Dial. 2019;23(6):507-515. PMID: 30498037
-
Hie M, et al. Rituximab in thrombotic thrombocytopenic purpura: a systematic review of the literature. Autoimmun Rev. 2020;19(10):102650. PMID: 32909630
-
Westwood JP, et al. Rituximab prophylaxis to prevent thrombotic thrombocytopenic purpura relapse: outcome and evaluation of dosing regimens. Blood. 2017;130(15):1746-1751. PMID: 28835437
-
Patriquin CJ, et al. Bortezomib in the treatment of refractory thrombotic thrombocytopenic purpura. Br J Haematol. 2016;173(5):779-785. PMID: 26992003
-
Sayani FA, et al. Platelet transfusion in TTP: harmful or helpful? Transfus Med Rev. 2016;30(3):96-102. PMID: 27056995
-
Fujimura Y, et al. Congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome): molecular pathogenesis, classification, diagnosis, and management. Semin Thromb Hemost. 2019;45(5):447-457. PMID: 30952310
-
Roose E, et al. ADAMTS13 and von Willebrand factor assessment in the diagnosis of thrombotic thrombocytopenic purpura and monitoring of patients in clinical remission. J Thromb Haemost. 2021;19(5):1158-1170. PMID: 33945620
Frequently asked questions
Quick clarifications for common clinical and exam-facing questions.
When should I seek emergency care for thrombotic thrombocytopenic purpura (adult)?
Seek immediate emergency care if you experience any of the following warning signs: Severe thrombocytopenia (less than 30 x10⁹/L) with schistocytes, Fluctuating neurological symptoms (confusion, seizures, stroke), Cardiac ischaemia with elevated troponin, Refractory to plasma exchange or PEX-dependent, Pregnancy with microangiopathic haemolytic anaemia, PLASMIC score >=5 (high probability TTP).
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Haemolytic Anaemia Overview
- Coagulation Cascade
Differentials
Competing diagnoses and look-alikes to compare.