Wilson Disease
The hallmark of WD is impaired biliary copper excretion and defective incorporation of copper into ceruloplasmin, resulting in toxic accumulation of free copper. Clinical presentations are highly heterogeneous,...
What matters first
The hallmark of WD is impaired biliary copper excretion and defective incorporation of copper into ceruloplasmin, resulting in toxic accumulation of free copper. Clinical presentations are highly heterogeneous,...
Acute liver failure (Wilsonian crisis)
6 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Acute liver failure (Wilsonian crisis)
- Coombs-negative haemolytic anaemia
- Young patient with unexplained cirrhosis
- Neuropsychiatric symptoms with liver disease
Linked comparisons
Differentials and adjacent topics worth opening next.
- Autoimmune Hepatitis
- Parkinson Disease
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Wilson Disease
1. Clinical Overview
Summary
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene on chromosome 13q14.3, leading to pathological copper accumulation predominantly in the liver, brain, cornea, and kidneys. The condition affects approximately 1 in 30,000 individuals worldwide, with clinical presentation typically occurring between ages 3-40 years, though peak incidence varies by phenotype: hepatic presentations commonly emerge in childhood to adolescence (5-15 years), while neuropsychiatric manifestations typically manifest in young adults (15-30 years). [1]
The hallmark of WD is impaired biliary copper excretion and defective incorporation of copper into ceruloplasmin, resulting in toxic accumulation of free copper. Clinical presentations are highly heterogeneous, ranging from asymptomatic biochemical abnormalities detected during family screening to fulminant hepatic failure requiring emergency liver transplantation. The three main phenotypic categories are: (1) hepatic WD (40-50% of cases) presenting with acute hepatitis, chronic hepatitis, cirrhosis, or acute liver failure; (2) neuropsychiatric WD (40-50%) with movement disorders (tremor, dystonia, parkinsonism), dysarthria, dysphagia, and psychiatric symptoms; and (3) presymptomatic WD detected through family screening. [2,3]
The pathognomonic ocular finding—Kayser-Fleischer (KF) rings—represents copper deposition in Descemet's membrane of the cornea and appears as golden-brown or greenish discolouration at the corneal periphery. KF rings are present in approximately 95% of patients with neurological WD but only 50-60% of those with purely hepatic presentations, emphasizing that their absence does not exclude the diagnosis. [4]
Early diagnosis and prompt initiation of lifelong copper chelation therapy (D-penicillamine, trientine) or zinc salts can halt disease progression, improve symptoms, and normalize life expectancy. Conversely, untreated WD is invariably fatal, progressing to end-stage liver disease, severe neurological disability, or both. Liver transplantation is curative for the metabolic defect and represents the only definitive treatment for acute liver failure or decompensated cirrhosis unresponsive to medical therapy. [5,6]
Key Facts
- Definition: Autosomal recessive disorder of copper metabolism due to ATP7B gene mutations
- Incidence: 1 in 30,000 live births; carrier frequency 1:90-1:100
- Peak Demographics: Presentation 3-40 years (hepatic: 5-15y; neurological: 15-30y)
- Genetics: > 500 pathogenic variants identified; H1069Q most common in Europeans
- Pathognomonic Features: Kayser-Fleischer rings + low ceruloplasmin + elevated urinary copper
- Gold Standard Diagnosis: Leipzig Score ≥4 (integrates clinical, biochemical, genetic data)
- First-line Treatment: D-penicillamine or trientine (chelation) ± zinc acetate
- Prognosis: Normal life expectancy if treated early; fatal if untreated
- Emergency: Fulminant WD requires urgent transplant assessment (high mortality)
Clinical Pearls
Diagnostic Pearl: The triad of low ceruloplasmin (less than 0.2 g/L), elevated 24-hour urinary copper (> 100 mcg/day), and Kayser-Fleischer rings on slit-lamp examination is virtually pathognomonic for Wilson disease. However, ceruloplasmin can be normal in 5-10% of cases, particularly in heterozygotes or those with acute inflammation.
Emergency Pearl: Fulminant Wilsonian crisis presents as acute liver failure with Coombs-negative haemolytic anaemia, disproportionately elevated AST/ALT ratio (> 2.2), low alkaline phosphatase, and very high serum copper. This constellation should trigger immediate transplant referral—medical therapy alone is inadequate.
Treatment Paradox Pearl: Up to 50% of patients with neurological WD experience transient worsening of neurological symptoms ("neurological deterioration paradox") during the first 3-6 months of D-penicillamine therapy. This represents mobilisation of hepatic copper stores and does NOT indicate treatment failure. Consider gradual dose escalation or switching to trientine.
Screening Pearl: All first-degree relatives of a confirmed WD patient should undergo screening (ceruloplasmin, 24h urinary copper, slit-lamp examination) regardless of age or symptoms. Siblings have a 25% risk of being affected; early detection in presymptomatic individuals dramatically improves outcomes.
Why This Matters Clinically
Wilson disease represents one of the few genuinely treatable inherited metabolic disorders. Early diagnosis and initiation of lifelong therapy can prevent irreversible hepatic and neurological damage, normalize life expectancy, and allow patients to lead normal lives. Conversely, delayed or missed diagnosis results in progressive disability and premature death. The condition must be considered in ALL young patients (less than 40 years) presenting with unexplained liver disease, movement disorders, psychiatric symptoms, or combinations thereof. Family screening is mandatory to detect presymptomatic cases.
2. Epidemiology
Incidence and Prevalence
| Parameter | Value | Notes |
|---|---|---|
| Global Incidence | 1 in 30,000 live births | Range: 1:10,000 to 1:100,000 depending on population [1] |
| Carrier Frequency | 1 in 90-100 | Implies heterozygote prevalence ~1.1% [7] |
| Male:Female Ratio | 1:1 | Equal sex distribution [2] |
| Prevalence | ~3-5 per 100,000 | Higher in populations with consanguinity [1] |
Exam Detail: Ethnic and Geographic Variations:
- Higher prevalence in certain populations due to founder effects and consanguinity
- Sardinia: 1 in 2,500 (founder mutation)
- Costa Rica: 1 in 15,000
- Japan and Korea: Similar to global average
- China: Some regions report higher prevalence (1 in 10,000)
- Ashkenazi Jews: No significantly elevated risk
The H1069Q mutation accounts for 30-70% of ATP7B alleles in European populations but is rare in Asian populations, which have distinct mutation spectra. [7,8]
Age of Presentation
| Phenotype | Typical Age Range | Peak Age | Notes |
|---|---|---|---|
| Hepatic WD | 5-35 years | 10-15 years | Can present in early childhood or later |
| Neurological WD | 12-40 years | 20-30 years | Rarely before age 12 |
| Psychiatric WD | 15-40 years | 20-30 years | Often co-exists with neurological features |
| Presymptomatic | Any age | — | Detected through family screening |
- Rare late presentations: Occasional cases diagnosed > 40 years, even in 6th-7th decade [9]
- Paediatric presentations: Can present as young as 3 years with hepatic disease
- Pregnancy: May unmask subclinical disease due to increased metabolic demands
Demographics
- Sex: Equal distribution in males and females
- Ethnicity: Affects all ethnic groups; mutation spectrum varies by population
- Family History:
- 25% sibling recurrence risk (autosomal recessive)
- Positive family history in ~20-30% of index cases
- Remaining cases represent new mutations or incomplete family histories
3. Aetiology and Pathophysiology
Genetic Basis
Exam Detail: ATP7B Gene:
- Located on chromosome 13q14.3
- Encodes a P-type ATPase copper transporter (1,465 amino acids)
- Predominantly expressed in hepatocytes
- Function: Transports copper from cytoplasm into trans-Golgi network for:
- Incorporation into ceruloplasmin (apoceruloplasmin → holoceruloplasmin)
- Excretion into bile (primary route of copper elimination)
Mutation Spectrum:
-
500 pathogenic variants identified worldwide [7]
- Missense mutations (60%), frameshift/nonsense (20%), splice-site (15%), deletions (5%)
- Most patients are compound heterozygotes (two different mutations)
- Genotype-phenotype correlation is weak and unreliable for predicting clinical course
Common Mutations by Population:
- H1069Q: 30-70% of European alleles (exon 14 missense)
- R778L: Common in Asian populations
- 3402delC: Frequent in Sardinia (founder effect)
- Absence of two identified mutations does NOT exclude diagnosis (some variants in regulatory regions not routinely sequenced)
Normal Copper Homeostasis
Copper Balance:
- Daily dietary intake: 1-2 mg
- Absorption: Proximal small intestine (50-60% absorbed)
- Total body copper: 50-120 mg
- Liver copper concentration (normal): less than 50 mcg/g dry weight
Physiological Copper Handling:
- Absorption: Intestinal enterocytes via CTR1 transporter
- Portal Transport: Bound to albumin and amino acids
- Hepatic Uptake: CTR1-mediated entry into hepatocytes
- Intracellular Trafficking: Chaperone proteins (ATOX1, CCS, COX17)
- Ceruloplasmin Synthesis: ATP7B incorporates copper into apoceruloplasmin in Golgi
- Biliary Excretion: ATP7B transports copper to canalicular membrane for bile secretion (1-2 mg/day)
Pathophysiology of Wilson Disease
Step 1: Loss of ATP7B Function
Biallelic ATP7B mutations result in:
- Impaired copper incorporation into ceruloplasmin → low serum ceruloplasmin
- Defective biliary copper excretion → hepatic copper accumulation
Exam Detail: Ceruloplasmin Biochemistry:
- Apoceruloplasmin (copper-free) is unstable and rapidly degraded
- Holoceruloplasmin (copper-loaded) has half-life of 5-6 days
- In WD: Low/absent holoceruloplasmin → low serum ceruloplasmin (less than 0.2 g/L in 90%)
- Note: Ceruloplasmin is an acute-phase reactant; can be falsely "normal" during inflammation, pregnancy, estrogen therapy
Why Copper Accumulates:
- Biliary excretion is the ONLY significant route of copper elimination (urinary excretion minimal)
- Without functional ATP7B, copper cannot reach bile canaliculi
- Dietary copper (1-2 mg/day) continuously accumulates in hepatocytes
Step 2: Hepatic Copper Overload
- Copper accumulates initially in hepatocyte lysosomes
- Liver copper content: > 250 mcg/g dry weight (normal: less than 50 mcg/g)
- Hepatic capacity eventually exceeded (typically after years)
Step 3: Non-Ceruloplasmin-Bound Copper Release
- Once hepatic storage capacity saturated, copper released into circulation as "free" (non-ceruloplasmin-bound) copper
- Free copper is highly toxic (pro-oxidant, generates reactive oxygen species)
- Deposits in extrahepatic tissues: brain, cornea, kidneys, heart
Calculation of Free Copper:
Free Copper (μg/dL) = Total Serum Copper – (Ceruloplasmin × 3)
[Normal: less than 10 μg/dL; WD: typically > 25 μg/dL]
Step 4: Organ-Specific Damage
| Organ | Mechanism of Injury | Clinical Manifestations |
|---|---|---|
| Liver | Oxidative stress, lipid peroxidation, mitochondrial dysfunction, apoptosis | Steatosis → hepatitis → fibrosis → cirrhosis; acute necrosis (fulminant) |
| Brain | Copper deposition in basal ganglia (putamen, globus pallidus, caudate), thalamus, brainstem; neuronal injury, astrocyte dysfunction, blood-brain barrier disruption | Movement disorders, cognitive impairment, psychiatric symptoms |
| Cornea | Copper deposition in Descemet's membrane | Kayser-Fleischer rings, sunflower cataracts |
| Kidney | Proximal tubule damage | Fanconi syndrome, nephrolithiasis, microscopic haematuria |
| Red Blood Cells | Copper-induced oxidative damage to RBC membranes | Coombs-negative haemolytic anaemia (especially in acute liver failure) |
| Heart | Myocardial copper deposition | Cardiomyopathy, arrhythmias (rare) |
| Bone | Copper deposition in joints | Arthropathy, osteoarthritis, osteoporosis |
Exam Detail: Neurological Pathology:
- Basal Ganglia: Bilateral putaminal and lenticular necrosis, cavitation
- Thalamus: Atrophy, gliosis
- Brainstem: Pontine and mesencephalic atrophy
- Cerebral Cortex: Usually spared until late stages
- Histology: Neuronal loss, Alzheimer type II astrocytosis, Opalski cells (large astrocytes with glycogen inclusions)
Hepatic Pathology:
- Early: Steatosis (macro and microvesicular), glycogenated nuclei
- Intermediate: Portal and periportal inflammation, interface hepatitis, fibrosis
- Advanced: Cirrhosis (macro or micronodular), regenerative nodules
- Acute: Massive hepatic necrosis, bridging necrosis
- Histochemistry: Rhodanine stain highlights copper in hepatocytes; orcein stain for copper-associated protein
4. Clinical Presentation
Wilson disease is a "great mimicker" with protean manifestations. Clinical presentation depends on age, primary organ involvement, and disease duration.
Hepatic Presentations (40-50% of cases)
| Presentation | Features | Typical Age | Frequency |
|---|---|---|---|
| Asymptomatic Transaminitis | Incidental finding of elevated ALT/AST; patient well | 5-20 years | 30-40% |
| Acute Hepatitis | Jaundice, fatigue, anorexia; mimics viral/autoimmune hepatitis | 5-30 years | 20-30% |
| Chronic Hepatitis | Persistent transaminitis, gradual progression to fibrosis | 5-35 years | 20-30% |
| Cirrhosis | Compensated or decompensated; portal hypertension, ascites | 10-40 years | 30-40% |
| Fulminant Hepatic Failure | Acute liver failure + haemolysis; high mortality | 8-35 years | 5-10% |
Exam Detail: Fulminant Wilsonian Crisis (Medical Emergency):
- Acute liver failure with jaundice, coagulopathy, encephalopathy
- Coombs-negative haemolytic anaemia (pathognomonic combination)
- Very high serum copper (> 200 mcg/dL) and urinary copper (> 1000 mcg/24h)
- Low alkaline phosphatase (less than 40 U/L) due to hepatocyte necrosis
- AST/ALT ratio typically > 2.2 (ratio > 4 highly suggestive) [10]
- Alkaline phosphatase/total bilirubin ratio less than 4 is 94% specific for Wilsonian crisis
- Rapid progression; medical therapy inadequate → emergency liver transplantation
- Mortality > 90% without transplantation
King's College Criteria (Modified for Wilson Disease): Not specifically validated for WD, but transplant should be considered when:
- INR > 2.0
- Rising bilirubin despite treatment
- Progressive encephalopathy
- Renal failure
New Wilson Index (Nazer Score): Predicts need for transplantation; score ≥11 indicates poor prognosis without transplant:
- Bilirubin (mg/dL) × 1 point
- INR × 10 points
- AST (U/L) × 0.01 points
- WBC (×10³/μL) × 1 point
- Albumin (g/dL) × (-1) points [Score ≥11: sensitivity 93%, specificity 98% for death without transplant] [11]
Neuropsychiatric Presentations (40-50% of cases)
Neurological Features:
| Symptom | Description | Frequency |
|---|---|---|
| Tremor | Rest, postural, or intention; "wing-beating" tremor (coarse, proximal, flapping with arms extended) | 50-65% |
| Dystonia | Focal or generalized; often affects face (risus sardonicus), neck, limbs | 45-60% |
| Dysarthria | Slurred speech, hypophonia, monotone; often earliest symptom | 90-97% |
| Dysphagia | Difficulty swallowing; drooling | 30-40% |
| Parkinsonism | Bradykinesia, rigidity, mask-like facies, gait disturbance | 25-40% |
| Ataxia | Cerebellar signs; truncal and limb ataxia | 30-40% |
| Chorea | Less common than dystonia | 10-15% |
| Seizures | Rare | 5-10% |
Psychiatric Features (present in 30-100% of neurological WD):
| Symptom | Description | Notes |
|---|---|---|
| Behavioural Changes | Irritability, aggression, impulsivity, disinhibition | Often earliest feature; may precede motor symptoms by years |
| Depression | Low mood, anhedonia, suicidal ideation | Most common psychiatric manifestation (30-40%) |
| Anxiety | Generalized anxiety, panic attacks | 20-30% |
| Psychosis | Hallucinations, delusions, paranoia | 10-15%; may mimic schizophrenia |
| Cognitive Impairment | Executive dysfunction, memory problems, reduced processing speed | Subcortical dementia pattern |
| Personality Change | Apathy, social withdrawal, reduced emotional expression | Often subtle; noticed by family |
Exam Detail: Neurological Examination Findings:
- Tremor: Best elicited with arms outstretched (wing-beating) or with finger-nose testing
- Dystonia: Facial grimacing, torticollis, limb dystonia; may be task-specific
- Dysarthria: Scanning speech, hypophonia, monotone, drooling
- Parkinsonism: Cogwheel rigidity, bradykinesia, reduced facial expression, shuffling gait
- Gait: Wide-based, unsteady, slow, reduced arm swing
- Reflexes: Variable; may be normal, brisk, or reduced
- Kayser-Fleischer rings: Visible with slit-lamp in 95% of neurological WD (see below)
"Flapping Tremor" Misnomer: The classic "wing-beating tremor" is NOT asterixis (which is a metabolic tremor seen in hepatic encephalopathy). Wing-beating tremor in WD is a proximal, rhythmic, coarse tremor elicited by shoulder abduction and elbow flexion, resembling a bird flapping wings.
Ophthalmological Features
Kayser-Fleischer (KF) Rings:
- Golden-brown or greenish copper deposition in Descemet's membrane (peripheral cornea)
- Begins at superior cornea, then inferior, eventually forms complete ring
- Slit-lamp examination required for definitive detection (not visible to naked eye in early stages)
- Prevalence:
- "Neurological WD: 95-100%"
- "Hepatic WD: 50-65%"
- "Presymptomatic WD: 20-30%"
- Absence does NOT exclude WD, especially in pure hepatic presentations
- KF rings may fade or disappear with successful chelation therapy
Sunflower Cataracts:
- Copper deposition in lens capsule
- Radial, petal-like pattern (resembles sunflower)
- Less common than KF rings (10-20%)
- Does not affect vision
- More common in chronic WD
Other Systemic Features
| System | Manifestations |
|---|---|
| Haematological | Coombs-negative haemolytic anaemia (especially acute presentations); thrombocytopenia (cirrhosis/portal hypertension) |
| Renal | Fanconi syndrome (proximal tubular dysfunction: glycosuria, aminoaciduria, phosphaturia, RTA type 2); nephrolithiasis; microscopic haematuria |
| Musculoskeletal | Arthropathy (knees, wrists, spine); osteoarthritis; osteoporosis; rickets/osteomalacia (Fanconi syndrome) |
| Cardiac | Cardiomyopathy (rare); arrhythmias; autonomic dysfunction |
| Dermatological | Hyperpigmentation (shins); blue lunulae (nails) |
| Endocrine | Hypoparathyroidism; amenorrhoea; delayed puberty; infertility (rare) |
Patterns of Presentation
Exam Detail: Phenotypic Categories [2]:
- Hepatic predominant: Liver disease ± minimal neurological signs; younger age
- Neurological predominant: Movement disorder ± mild liver disease; older age
- Mixed hepatoneurological: Both hepatic and neurological features
- Presymptomatic: Detected via family screening; normal examination or subtle findings
- Rare presentations: Isolated haemolysis, renal disease, cardiac involvement
Age-Related Presentation Patterns:
- Children (3-10 years): Almost exclusively hepatic; often asymptomatic transaminitis
- Adolescents (10-20 years): Hepatic or mixed; increasing neuropsychiatric features
- Young Adults (20-30 years): Predominantly neuropsychiatric; often with established cirrhosis
- Adults (> 30 years): Variable; late diagnosis often involves neurological or psychiatric manifestations
Red Flags (Emergency Features)
[!CAUTION] Red Flags Requiring Urgent Assessment
- Young patient (less than 40 years) with unexplained cirrhosis → Screen for WD
- Acute liver failure + Coombs-negative haemolysis → Fulminant WD; emergency transplant referral
- Movement disorder + liver disease in young adult → WD until proven otherwise
- AST/ALT ratio > 2.2 with acute hepatitis → Suggests Wilsonian crisis
- Alkaline phosphatase/bilirubin less than 4 in acute liver failure → Highly specific for WD
- Kayser-Fleischer rings in patient with neuropsychiatric symptoms → Virtually diagnostic
- Family history of unexplained liver disease or young-onset movement disorder → Screen for WD
5. Differential Diagnosis
Wilson disease must be distinguished from other causes of liver disease, movement disorders, and psychiatric illness.
Hepatic Presentations
| Condition | Distinguishing Features | Key Investigations |
|---|---|---|
| Autoimmune Hepatitis | Female predominance; ANA, ASMA, anti-LKM positive; hypergammaglobulinaemia | Autoantibodies, IgG, liver biopsy |
| Viral Hepatitis (B, C) | Risk factors (IVDU, transfusion); positive serology | HBsAg, anti-HCV, HCV RNA |
| Non-Alcoholic Fatty Liver Disease | Metabolic syndrome; steatosis on imaging | Ultrasound, liver biopsy |
| Alcoholic Liver Disease | Alcohol history; AST>ALT; macrocytosis | History, GGT, AST/ALT ratio |
| Hereditary Haemochromatosis | Elevated ferritin, transferrin saturation > 45%; HFE mutation | Ferritin, transferrin saturation, HFE gene |
| Alpha-1 Antitrypsin Deficiency | Low AAT level; PiZZ genotype; lung disease | AAT level, phenotype, gene testing |
| Budd-Chiari Syndrome | Hepatomegaly, ascites; IVC/hepatic vein thrombosis on imaging | Doppler ultrasound, CT/MRI |
Comparison Table: Wilson Disease vs Autoimmune Hepatitis
| Feature | Wilson Disease | Autoimmune Hepatitis |
|---|---|---|
| Age | 5-40 years | Bimodal: 10-20y, 50-60y |
| Sex | Equal | Female 70-80% |
| Ceruloplasmin | Low (less than 0.2 g/L) | Normal |
| Autoantibodies | Negative | ANA, ASMA, anti-LKM positive |
| IgG | Normal | Elevated |
| KF Rings | Present 50-95% | Absent |
| Urinary Copper | Elevated | Normal |
| Treatment | Chelation/zinc | Immunosuppression |
Neurological Presentations
| Condition | Distinguishing Features | Key Investigations |
|---|---|---|
| Parkinson Disease | Older age (> 50); asymmetric onset; levodopa-responsive; no KF rings | Clinical; DaTscan normal in WD |
| Essential Tremor | Postural/action tremor; family history; alcohol-responsive; no other features | Clinical; no KF rings or liver disease |
| Huntington Disease | Chorea predominant; positive family history; psychiatric features | HTT gene CAG repeat expansion |
| Multiple Sclerosis | Relapsing-remitting; white matter lesions; optic neuritis | MRI brain/spine; CSF oligoclonal bands |
| Dystonic Syndromes | Focal onset; no liver/eye signs | Clinical; genetic testing (e.g., DYT1) |
| Neurodegeneration with Brain Iron Accumulation | Progressive; "eye of the tiger" sign on MRI | MRI; gene panel (PANK2, etc.) |
Comparison Table: Wilson Disease vs Parkinson Disease
| Feature | Wilson Disease | Parkinson Disease |
|---|---|---|
| Age of Onset | 15-30 years | > 50 years (typically) |
| Tremor | Rest, postural, wing-beating | Rest tremor (pill-rolling) |
| Symmetry | Often symmetric | Asymmetric onset |
| Dysarthria | Common, early, severe | Late feature |
| Dystonia | Common | Uncommon |
| Liver Disease | Present | Absent |
| KF Rings | Present 95% | Absent |
| MRI Brain | Basal ganglia T2 hyperintensity | Normal (DaTscan reduced uptake) |
| Levodopa Response | Poor/nil | Excellent (initially) |
Psychiatric Presentations
| Condition | Distinguishing Features | Key Investigations |
|---|---|---|
| Schizophrenia | Earlier onset; no movement disorder (unless drug-induced); no liver/eye signs | Clinical; brain imaging normal |
| Bipolar Disorder | Episodic mood swings; no liver/neurological signs | Clinical |
| Major Depressive Disorder | Isolated mood symptoms; no movement disorder | Clinical |
| Substance-Induced Psychosis | Toxicology positive; temporal relationship | Urine drug screen |
| Frontotemporal Dementia | Older age; progressive behavioural change; temporal/frontal atrophy | MRI, neuropsychology |
Key Principle: Always screen for WD in young patients (less than 40 years) with new-onset psychiatric symptoms, especially if accompanied by movement disorder, liver disease, or family history.
6. Investigations
Screening Tests
| Test | Normal Range | Wilson Disease Finding | Sensitivity | Specificity |
|---|---|---|---|---|
| Serum Ceruloplasmin | 0.2-0.4 g/L (20-40 mg/dL) | less than 0.2 g/L (less than 20 mg/dL) | 85-95% | 60-70% |
| 24-Hour Urinary Copper | less than 40 mcg/day | > 100 mcg/day (untreated) | 75-95% | 80-95% |
| Slit-Lamp Examination | No KF rings | KF rings present | 50-95% (phenotype-dependent) | ~100% (age less than 10) |
Exam Detail: Ceruloplasmin Interpretation:
- Low (less than 0.2 g/L): Seen in 90% of WD, but also in:
- Heterozygous carriers (10-20% have low-normal ceruloplasmin)
- Severe liver disease (any cause)
- Nephrotic syndrome
- Protein-losing enteropathy
- Malnutrition
- Aceruloplasminaemia (rare genetic disorder)
- Normal (0.2-0.4 g/L): Does NOT exclude WD (5-10% of patients have "normal" ceruloplasmin)
- Acute inflammation (ceruloplasmin is acute-phase reactant)
- Pregnancy, estrogen therapy
- Heterozygotes
24-Hour Urinary Copper Interpretation:
- Normal (less than 40 mcg/day): Unlikely to be WD (but can occur in presymptomatic patients)
- Borderline (40-100 mcg/day): Suggestive; consider repeat, d-penicillamine challenge
- Elevated (> 100 mcg/day): Highly suggestive of WD
- Very High (> 1000 mcg/day): Seen in fulminant WD
- False positives: Cholestatic liver disease, chronic active hepatitis, nephrotic syndrome
Penicillamine Challenge Test (if urinary copper borderline):
- Administer 500 mg D-penicillamine PO at 0h and 12h
- Collect urine 0-24h for copper
- Interpretation: > 25-fold increase or total > 1600 mcg/24h strongly suggests WD
- Use limited; genetic testing now preferred
Advanced Biochemical Tests
| Test | Finding in WD | Interpretation |
|---|---|---|
| Serum Free (Non-Ceruloplasmin-Bound) Copper | > 25 mcg/dL (> 1.6 μmol/L) | Calculated: Total Cu – (Ceruloplasmin × 3); elevated in WD |
| Serum Total Copper | Often low or normal | Total copper = ceruloplasmin-bound + free; low due to low ceruloplasmin |
| Liver Copper Content | > 250 mcg/g dry weight | Gold standard if biopsy feasible; > 250 mcg/g diagnostic |
| Coombs Test | Negative (despite haemolysis) | Coombs-negative haemolysis in fulminant WD |
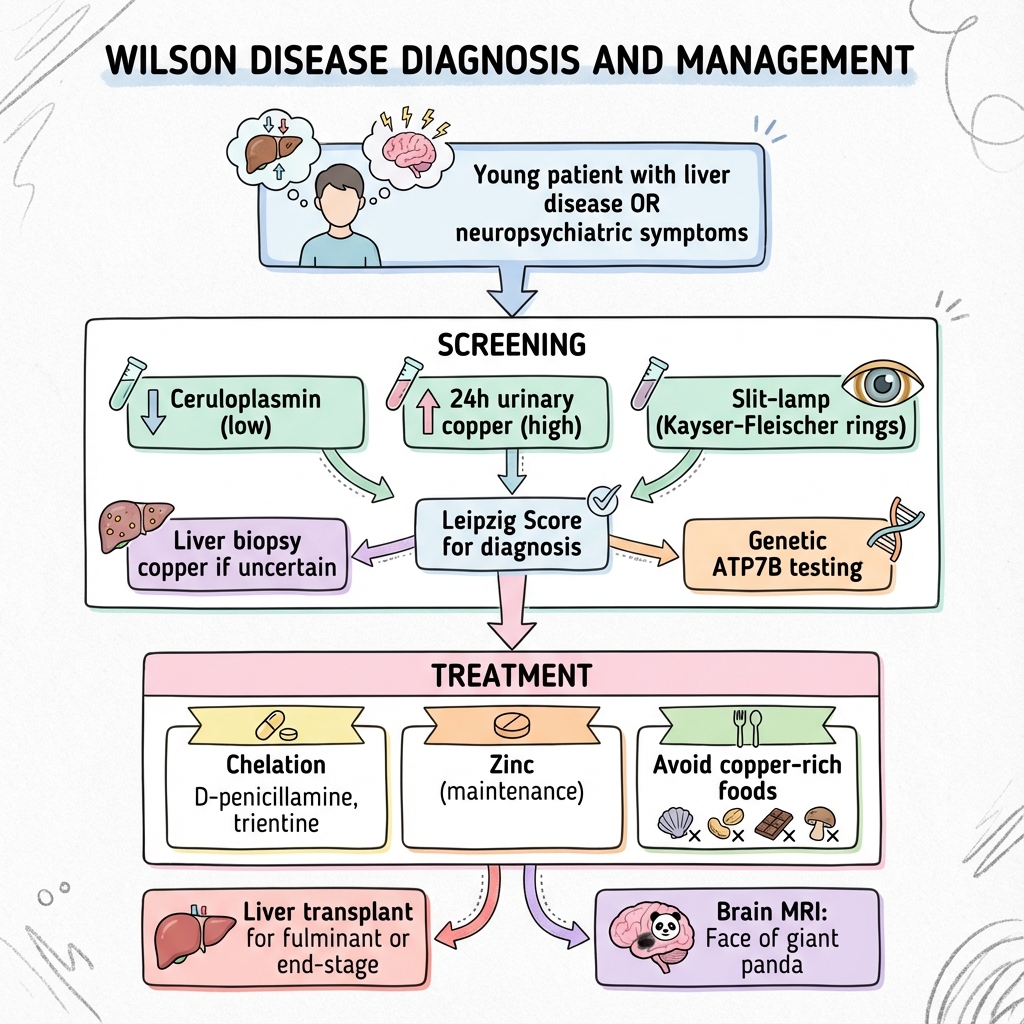
Leipzig Scoring System
The Leipzig Score integrates clinical, biochemical, and genetic data to diagnose WD. [12]
| Parameter | Finding | Points |
|---|---|---|
| Kayser-Fleischer Rings | Present | 2 |
| Absent | 0 | |
| Neuropsychiatric Symptoms | Severe (significant impairment) | 2 |
| Mild (subtle signs) | 1 | |
| Absent | 0 | |
| Coombs-Negative Haemolytic Anaemia | Present | 1 |
| Absent | 0 | |
| Serum Ceruloplasmin | Normal (≥0.2 g/L) | 0 |
| 0.1-0.2 g/L | 1 | |
| less than 0.1 g/L | 2 | |
| 24h Urinary Copper | Normal (less than 40 mcg) | 0 |
| 40-80 mcg | 1 | |
| 80-100 mcg | 2 | |
| > 100 mcg (or > 1600 mcg post-penicillamine) | 2 | |
| Liver Copper Content | Normal (less than 50 mcg/g) | -1 |
| 50-250 mcg/g | 1 | |
| > 250 mcg/g | 2 | |
| Rhodanine-Positive Granules | Present | 1 |
| (on liver histology, if no copper quantification) | Absent | 0 |
| ATP7B Mutations | 2 pathogenic mutations detected | 4 |
| 1 pathogenic mutation detected | 1 | |
| No mutations detected | 0 |
Interpretation:
- Score ≥4: Diagnosis of WD established
- Score 3: Diagnosis possible; further investigations required
- Score ≤2: Diagnosis unlikely (but not excluded; consider repeat testing, genetic studies)
Exam Detail: Advantages of Leipzig Score:
- Quantitative, reproducible
- Integrates multiple diagnostic parameters
- Useful when individual tests equivocal
- Validated in large cohorts
Limitations:
- Requires complete data set (often liver biopsy and genetic testing not immediately available)
- Genetic testing may not identify all mutations (novel variants, intronic mutations)
- Does not replace clinical judgment
Imaging
MRI Brain (T2-weighted, FLAIR sequences):
| Finding | Description | Frequency in Neurological WD |
|---|---|---|
| Basal Ganglia Hyperintensity | Bilateral symmetric T2/FLAIR hyperintensity in putamen, globus pallidus, caudate | 90-100% |
| "Face of the Giant Panda" Sign | Hyperintensity of superior colliculi, hypointensity of substantia nigra, tegmentum hyperintensity (midbrain) | 15-20% (specific for WD) |
| "Miniature Sunflower" Sign | Pontine hyperintensity | Less common |
| Thalamic Hyperintensity | Bilateral symmetric | 50-70% |
| White Matter Changes | Periventricular hyperintensity | 30-40% |
| Cortical Atrophy | Cerebral, cerebellar atrophy (late) | Variable |
Liver Imaging (Ultrasound, CT, MRI):
- Variable findings: normal, steatosis, hepatomegaly, cirrhosis, nodular liver
- Not specific for WD
- Used to assess liver architecture, portal hypertension, exclude HCC
Abdominal Ultrasound:
- Assess liver size, echogenicity, nodularity
- Evaluate spleen (splenomegaly suggests portal hypertension)
- Assess for ascites
Liver Biopsy
Indications:
- Diagnosis uncertain based on non-invasive tests
- Assess severity of liver disease (fibrosis staging)
- Quantitative hepatic copper measurement (gold standard)
Findings:
- Hepatic Copper Content: > 250 mcg/g dry weight (normal: less than 50 mcg/g) diagnostic
- Histology:
- Steatosis (macro/microvesicular)
- Portal/periportal inflammation
- Interface hepatitis (resembles autoimmune hepatitis)
- Fibrosis (variable stages)
- Cirrhosis (advanced cases)
- Glycogenated nuclei
- Mallory-Denk bodies (rare)
- Histochemistry:
- "Rhodanine stain: Highlights copper deposits (reddish granules)"
- "Orcein stain: Copper-associated protein (brown granules)"
Limitations:
- Copper distribution may be heterogeneous (sampling error)
- Elevated copper also seen in cholestatic liver diseases
- Invasive procedure with risks
Genetic Testing
ATP7B Gene Sequencing:
- Detects > 95% of pathogenic mutations in coding regions
- Confirms diagnosis if 2 pathogenic variants identified
- Useful for family screening, prenatal diagnosis
- Negative result does NOT exclude WD (intronic/regulatory mutations may not be detected)
Approach:
- Targeted mutation analysis (if specific population/family mutation known)
- Full gene sequencing (exons + splice sites)
- Deletion/duplication analysis if sequencing negative
Screening First-Degree Relatives
Recommended Protocol [6]:
- Serum ceruloplasmin (low in 90% of affected)
- 24-hour urinary copper (elevated in affected)
- Slit-lamp examination (KF rings in presymptomatic individuals)
- ATP7B genetic testing (if index case mutations known)
- Liver function tests (ALT, AST, bilirubin)
Interpretation:
- If all tests normal → unlikely to be affected (but repeat in 1-2 years if young)
- If abnormal → proceed to full diagnostic workup
7. Management
Wilson disease requires lifelong treatment. Goals: reduce copper accumulation, prevent disease progression, improve symptoms.
General Principles
- Lifelong Therapy: Treatment must continue indefinitely; discontinuation leads to relapse and death
- Individualized Regimen: Choice of initial therapy depends on phenotype, severity, tolerance
- Monitoring: Regular biochemical, clinical, and ophthalmological follow-up mandatory
- Avoid Copper: Dietary restriction, avoid copper-containing supplements
- Family Screening: Screen all first-degree relatives
Pharmacological Treatment
Exam Detail: Treatment Algorithm:
Wilson Disease Diagnosis Confirmed
|
├─> Presymptomatic / Mild Hepatic
| └─> Zinc Acetate (First-line)
| OR Trientine (if zinc not tolerated)
|
├─> Symptomatic Hepatic (No Liver Failure)
| └─> D-Penicillamine OR Trientine
| (Start low, escalate gradually)
| Add Zinc after 3-6 months
|
├─> Neurological Presentation
| └─> Trientine (preferred) OR D-Penicillamine (low dose)
| CAUTION: Risk neurological worsening
| Add Zinc after 3-6 months
|
├─> Fulminant Hepatic Failure
| └─> Emergency Liver Transplantation
| (Chelation insufficient)
|
└─> Maintenance (After Stabilization)
└─> Zinc Monotherapy
OR Low-dose Chelator + Zinc
1. Chelation Therapy
D-Penicillamine (First-line chelator):
| Parameter | Details |
|---|---|
| Mechanism | Copper chelator; forms soluble complex excreted in urine |
| Dose | Initial: 250 mg OD, increase gradually to 1000-1500 mg/day (in divided doses, 2-4 times daily) |
| Timing | 30-60 min before meals (empty stomach enhances absorption) |
| Monitoring | FBC, urinalysis, 24h urinary copper (weekly initially, then monthly) |
| Efficacy | Highly effective; gold standard for decades |
| Adverse Effects | Common (20-30%): hypersensitivity, rash, fever, arthralgia, proteinuria, lupus-like syndrome; Rare: bone marrow suppression, nephrotic syndrome, Goodpasture syndrome |
| Neurological Worsening | 20-50% patients experience initial worsening of neurological symptoms (first 3-6 months) |
| Contraindications | Penicillin allergy (relative); pregnancy (teratogenic—use with caution) |
| Adjuncts | Pyridoxine (vitamin B6) 25-50 mg/day to prevent deficiency |
Trientine (Triethylene tetramine dihydrochloride):
| Parameter | Details |
|---|---|
| Mechanism | Copper chelator (weaker than penicillamine); forms complex excreted in urine |
| Dose | 750-1500 mg/day (divided 2-3 times daily); children: 20 mg/kg/day |
| Timing | Empty stomach (30-60 min before meals) |
| Indications | Penicillamine intolerance; neurological patients (lower risk worsening than penicillamine) |
| Efficacy | Effective; often preferred in neurological WD |
| Adverse Effects | Generally better tolerated than penicillamine; sideroblastic anaemia (rare), gastritis, rash |
| Neurological Worsening | Lower risk than penicillamine (~10-20%) |
Exam Detail: Neurological Worsening Paradox:
- 20-50% of patients with neurological WD experience worsening of neurological symptoms during initial 3-6 months of chelation (especially D-penicillamine)
- Mechanism: Rapid mobilization of hepatic copper → transient increase in free serum copper → brain deposition
- Clinical features: Worsening tremor, dystonia, dysarthria, gait instability
- Management:
- Reassure patient (usually transient, resolves after 6-12 months)
- Continue treatment (discontinuation worsens outcome)
- Consider dose reduction, slow escalation
- Switch to trientine if severe worsening
- Add zinc acetate
- Prevention: Start with low doses, gradual escalation; consider trientine as first-line in neurological patients
2. Zinc Therapy
Zinc Acetate (or Zinc Sulfate):
| Parameter | Details |
|---|---|
| Mechanism | Induces metallothionein in intestinal enterocytes → binds dietary copper → blocks absorption; promotes fecal copper excretion |
| Dose | Adults: 50 mg elemental zinc TDS (150 mg/day); Children: 25 mg TDS |
| Timing | 1 hour before or 2 hours after meals (food reduces absorption) |
| Indications | Maintenance therapy; presymptomatic patients; pregnancy (safe); adjunct to chelation |
| Efficacy | Effective for maintenance; slower onset than chelators (not for acute treatment) |
| Adverse Effects | Gastric irritation, nausea (take with small snack if intolerable); rare: copper deficiency |
| Monitoring | 24h urinary zinc (aim > 2 mg/day confirms compliance); serum zinc; FBC |
| Advantages | Well-tolerated; no neurological worsening; safe in pregnancy |
Combination Therapy:
- Chelator (penicillamine or trientine) for initial de-coppering (6-12 months)
- Add zinc after 3-6 months
- Transition to zinc monotherapy for long-term maintenance (once stable)
3. Tetrathiomolybdate (Investigational)
| Parameter | Details |
|---|---|
| Mechanism | Forms tripartite complex (copper-molybdenum-protein) → prevents copper absorption and mobilization |
| Status | Approved in some countries (e.g., USA: bis-choline tetrathiomolybdate); investigational elsewhere |
| Indications | Neurological WD (may have lower risk of worsening than penicillamine) |
| Dose | Variable (clinical trials: 120-180 mg/day in divided doses) |
| Adverse Effects | Hepatotoxicity, bone marrow suppression, neurological toxicity |
| Monitoring | LFTs, FBC, neurological examination |
Liver Transplantation
Indications:
- Fulminant Hepatic Failure (Wilsonian Crisis):
- Acute liver failure + Coombs-negative haemolysis
- Medical therapy inadequate
- Emergency transplantation required (highest priority listing)
- Decompensated Cirrhosis:
- Refractory ascites, variceal bleeding, hepatorenal syndrome
- No improvement despite 3-6 months chelation therapy
- Hepatocellular Carcinoma (rare in WD but can occur)
Outcomes:
- 5-year survival: 80-90% [13]
- Curative: Corrects metabolic defect (donor liver has normal ATP7B)
- Neurological symptoms may improve post-transplant (but slower than hepatic features)
- Lifelong immunosuppression required
Prognostic Scores for Transplant Decision:
- New Wilson Index (Nazer Score): ≥11 predicts death without transplant
- King's College Criteria (adapted): Encephalopathy + INR > 2.0
- MELD Score: Standard liver transplant prioritization
Exam Detail: Transplant Controversies:
- Should liver transplantation be offered for isolated neurological WD refractory to medical therapy?
- Controversial; some centres offer transplant for severe, progressive neurological disease
- Evidence limited; neurological improvement variable
- Risk of immunosuppression vs. potential benefit
Dietary Management
Dietary Copper Restriction (especially during initial treatment):
| Food Group | Advice |
|---|---|
| Avoid (High Copper) | Shellfish (oysters, lobster, crab), liver, organ meats, chocolate, nuts (cashews, Brazil nuts), mushrooms, legumes (chickpeas, lentils), whole grains |
| Limit (Moderate Copper) | Beef, pork, dried fruit, avocado |
| Safe (Low Copper) | Dairy products, eggs, most vegetables, fruits, refined grains, chicken, fish (non-shellfish) |
Additional Advice:
- Avoid copper-containing multivitamins/supplements
- Check drinking water copper content (if copper pipes, may need filtration)
- Use stainless steel or non-copper cookware
Monitoring During Treatment
| Parameter | Frequency | Target/Interpretation |
|---|---|---|
| 24h Urinary Copper | Monthly (initial 3-6 months), then every 3-6 months | Initial: > 200 mcg/day (confirms decoppering); Maintenance: 50-150 mcg/day |
| Serum Free Copper | Every 3-6 months | Target: 5-15 mcg/dL |
| Serum Ceruloplasmin | Every 6-12 months | Usually remains low (not a monitoring parameter) |
| Liver Function Tests | Every 3-6 months | Monitor for hepatotoxicity, improvement |
| Full Blood Count | Every 3-6 months | Monitor for drug-induced cytopenias |
| Urinalysis | Every 3-6 months (if on penicillamine) | Proteinuria (penicillamine nephrotoxicity) |
| Slit-Lamp Examination | Annually | KF rings may fade/disappear with treatment |
| Neurological Examination | Every 3-6 months | Monitor symptom progression/improvement |
| Compliance Assessment | Every visit | Non-compliance common cause of relapse |
Treatment Targets:
- Clinical: Symptom stabilization or improvement
- Biochemical: 24h urinary copper 50-150 mcg/day (maintenance); free copper 5-15 mcg/dL
- Ophthalmological: Fading of KF rings (may take years)
Special Populations
Pregnancy [14]:
- Continue treatment throughout pregnancy (discontinuation risks maternal/fetal harm)
- Zinc acetate: Safest option; first-line in pregnancy
- D-penicillamine: Continue if already on (reduce dose to 750-1000 mg/day); associated with connective tissue abnormalities in fetus (rare); benefits usually outweigh risks
- Trientine: Limited data; probably safe; continue if already on
- Monitoring: More frequent monitoring (monthly)
- Delivery: No specific contraindications; vaginal delivery safe
Children:
- Doses adjusted for weight
- Zinc: 25 mg TDS
- Penicillamine/Trientine: 20 mg/kg/day (divided doses)
- Monitor growth, development
Elderly:
- Rare late presentations
- Adjust doses for renal function, comorbidities
- Monitor for drug interactions
Non-Compliance
- Major cause of treatment failure and relapse
- Factors: Lifelong therapy burden, side effects, lack of symptoms (presymptomatic patients)
- Consequences: Relapse of liver/neurological disease; acute liver failure
- Strategies: Education, regular follow-up, pill counts, urinary copper monitoring (low urinary copper suggests non-compliance)
8. Complications
Hepatic Complications
| Complication | Description | Management |
|---|---|---|
| Cirrhosis | Progressive fibrosis → portal hypertension | Surveillance for varices, HCC; transplant if decompensated |
| Portal Hypertension | Varices, ascites, splenomegaly | Variceal screening (endoscopy); beta-blockers; band ligation |
| Hepatocellular Carcinoma | Rare in WD (lower risk than viral/NASH cirrhosis) | Surveillance: 6-monthly ultrasound + AFP |
| Acute Liver Failure | Fulminant Wilsonian crisis | Emergency transplantation |
Neurological Complications
| Complication | Description | Management |
|---|---|---|
| Permanent Neurological Disability | Residual tremor, dystonia, dysarthria despite treatment | Multidisciplinary: neurology, speech therapy, occupational therapy, physiotherapy |
| Neurological Worsening on Treatment | Paradoxical deterioration (first 3-6 months) | Reassurance, continue treatment, consider dose reduction/switch to trientine |
| Cognitive Impairment | Executive dysfunction, memory problems | Cognitive rehabilitation, supportive care |
| Psychiatric Complications | Depression, anxiety, psychosis | Psychiatry input, antidepressants, antipsychotics |
Renal Complications
| Complication | Description | Management |
|---|---|---|
| Fanconi Syndrome | Proximal tubular dysfunction | Phosphate, bicarbonate, vitamin D supplementation |
| Nephrolithiasis | Renal stones (hypercalciuria) | Hydration, dietary modification, urology referral |
| Penicillamine Nephrotoxicity | Proteinuria, nephrotic syndrome | Switch to trientine/zinc |
Haematological Complications
| Complication | Description | Management |
|---|---|---|
| Haemolytic Anaemia | Coombs-negative haemolysis (fulminant WD) | Supportive care, transfusion, emergency transplant |
| Drug-Induced Cytopenias | Penicillamine bone marrow suppression | Stop penicillamine, switch to trientine/zinc; monitor FBC |
Skeletal Complications
| Complication | Description | Management |
|---|---|---|
| Osteoporosis | Bone loss (disease + penicillamine effect) | DEXA scan, calcium, vitamin D, bisphosphonates |
| Arthropathy | Joint pain, osteoarthritis | Analgesia, physiotherapy |
9. Prognosis
With Treatment
-
Early Diagnosis (Presymptomatic/Early Symptomatic):
- Excellent prognosis
- Normal life expectancy with adherence to therapy [6]
- Hepatic disease stabilizes or improves
- Neurological symptoms may stabilize (may not fully reverse)
- KF rings may fade or disappear
-
Symptomatic at Diagnosis:
- Variable outcomes depending on severity
- "Hepatic: Good response if pre-cirrhotic; slower improvement if cirrhotic"
- "Neurological: Improvement over months to years; residual disability common"
- Psychiatric symptoms often improve with treatment
-
Advanced Disease (Cirrhosis, Neurological Disability):
- Stabilization possible but reversal limited
- Ongoing monitoring for complications (varices, HCC)
- Quality of life depends on residual symptoms
Without Treatment
- Uniformly Fatal
- Progression to:
- End-stage liver disease (hepatic failure)
- Severe neurological disability (wheelchair-bound, anarthria)
- Death (usually hepatic or neurological deterioration)
- Median survival untreated: less than 5 years from symptom onset
Factors Affecting Prognosis
| Factor | Impact on Prognosis |
|---|---|
| Age at Diagnosis | Younger age (presymptomatic) → better outcomes |
| Phenotype at Presentation | Presymptomatic > hepatic > neurological (in terms of reversibility) |
| Severity at Diagnosis | Early disease → excellent prognosis; fulminant/advanced → worse |
| Treatment Adherence | Non-compliance → relapse, worse outcomes |
| Neurological Involvement | Neurological symptoms less likely to fully reverse than hepatic |
| Time to Treatment | Earlier treatment → better long-term outcomes |
Post-Transplant Prognosis
- 5-year survival: 80-90% [13]
- Curative for metabolic defect
- Neurological symptoms may improve (but slower than hepatic features)
- Lifelong immunosuppression required (associated risks)
10. Prevention and Screening
Primary Prevention
- No primary prevention (genetic disorder)
- Genetic counseling for families with known WD
Secondary Prevention (Early Detection)
Family Screening:
- All first-degree relatives (siblings, children) of index case should be screened [6]
- Screening Tests:
- Serum ceruloplasmin
- 24-hour urinary copper
- Slit-lamp examination
- Liver function tests
- ATP7B genetic testing (if index mutations known)
- Timing: As soon as diagnosis in index case confirmed; repeat in childhood if initially normal
- Outcome: Early treatment of presymptomatic individuals prevents disease manifestations
Cascade Screening:
- If sibling affected, screen their children (index case's nieces/nephews)
Tertiary Prevention (Prevent Complications)
- Lifelong treatment adherence: Prevent relapse
- Regular monitoring: Detect complications early (varices, HCC)
- Avoid hepatotoxins: Alcohol, hepatotoxic drugs
- Vaccination: Hepatitis A and B (protect liver)
- Surveillance:
- Variceal screening (endoscopy) if cirrhotic
- HCC surveillance (6-monthly ultrasound + AFP) if cirrhotic
11. Key Guidelines and Evidence
Major Guidelines
-
European Association for the Study of the Liver (EASL) 2012 [3]:
- Comprehensive diagnostic and management guidelines
- Leipzig Score for diagnosis
- Treatment recommendations (chelation, zinc, transplant)
-
American Association for the Study of Liver Diseases (AASLD) 2008 [1]:
- Diagnostic criteria
- Treatment algorithms
- Transplant indications
-
British Society of Gastroenterology (BSG) Guidelines (upcoming update expected)
Landmark Studies
- Walshe JS. Lancet 1956 [15]: First description of penicillamine treatment for WD
- Brewer GJ et al. Arch Neurol 1991 [16]: Zinc acetate efficacy in WD
- Ferenci P et al. Gastroenterology 2003 [12]: Development and validation of Leipzig Score
- Litwin T et al. Mov Disord 2012 [17]: Characterization of neurological worsening with chelation
12. Examination Focus
Viva Pearls
Opening Statement: "Wilson disease is an autosomal recessive disorder of copper metabolism caused by ATP7B gene mutations on chromosome 13, leading to impaired biliary copper excretion and defective ceruloplasmin synthesis. This results in toxic copper accumulation in the liver, brain, cornea, and other organs. It presents with hepatic disease, neuropsychiatric symptoms, or both, typically in young adults. Early diagnosis and lifelong copper chelation therapy are essential; untreated disease is fatal."
Common Viva Questions and Model Answers
Q1: How do you diagnose Wilson disease?
"Diagnosis is based on a combination of clinical features, biochemical tests, and genetic analysis. The Leipzig Score integrates these parameters. Key investigations include: (1) serum ceruloplasmin—low (less than 0.2 g/L) in 90% of cases, though can be normal; (2) 24-hour urinary copper—elevated (> 100 mcg/day) in untreated WD; (3) slit-lamp examination for Kayser-Fleischer rings—present in 95% of neurological WD and 50% of hepatic WD; (4) serum free copper—calculated as total copper minus ceruloplasmin-bound copper, elevated in WD; (5) liver copper content on biopsy (if feasible)—> 250 mcg/g dry weight is diagnostic; and (6) ATP7B genetic testing—detection of two pathogenic mutations confirms diagnosis. A Leipzig Score ≥4 establishes the diagnosis."
Q2: What is the pathophysiology of copper accumulation in Wilson disease?
"ATP7B is a copper-transporting ATPase located in hepatocytes. It has two critical functions: (1) incorporating copper into apoceruloplasmin in the Golgi to form holoceruloplasmin, and (2) transporting copper to the bile canaliculus for excretion into bile, which is the primary route of copper elimination. In WD, loss-of-function mutations in ATP7B impair both processes. This leads to reduced serum ceruloplasmin (apoceruloplasmin is unstable and degraded) and impaired biliary copper excretion. Consequently, copper accumulates in hepatocytes. Over time, hepatic storage capacity is exceeded, and copper is released into the bloodstream as toxic non-ceruloplasmin-bound 'free' copper, which deposits in extrahepatic tissues—particularly the brain (basal ganglia), cornea (Kayser-Fleischer rings), and kidneys—causing oxidative damage and organ dysfunction."
Q3: Describe the clinical presentations of Wilson disease.
"WD has three main phenotypes. (1) Hepatic: Ranges from asymptomatic transaminitis to acute hepatitis, chronic hepatitis, cirrhosis, or fulminant hepatic failure. Fulminant WD (Wilsonian crisis) presents with acute liver failure, Coombs-negative haemolytic anaemia, very high serum copper, and high AST/ALT ratio (> 2.2)—this is a medical emergency requiring transplant. (2) Neuropsychiatric: Movement disorders (tremor—often 'wing-beating', dystonia, parkinsonism), dysarthria, dysphagia, psychiatric symptoms (depression, behavioural changes, psychosis), and cognitive impairment. Kayser-Fleischer rings are present in 95% of neurological cases. (3) Presymptomatic: Detected via family screening; may have subtle biochemical or ophthalmological findings but no overt symptoms. Presentations typically occur age 5-40 years, with hepatic earlier (5-15y) and neurological later (15-30y)."
Q4: What are the treatment options for Wilson disease?
"Treatment aims to reduce copper accumulation and is lifelong. Options include: (1) Chelation therapy—D-penicillamine or trientine. Penicillamine (1000-1500 mg/day) is highly effective but has significant side effects (hypersensitivity, proteinuria, bone marrow suppression) and can cause neurological worsening in 20-50% of patients. Trientine (750-1500 mg/day) is better tolerated and preferred in neurological WD. (2) Zinc therapy—zinc acetate (150 mg elemental zinc/day) blocks intestinal copper absorption and is used for maintenance therapy or in presymptomatic patients. It's safe in pregnancy. (3) Combination therapy—initial chelation for de-coppering (6-12 months), then transition to zinc monotherapy or low-dose chelator plus zinc. (4) Liver transplantation—curative; indicated for fulminant hepatic failure or decompensated cirrhosis unresponsive to medical therapy. (5) Dietary copper restriction—avoid shellfish, liver, nuts, chocolate. Treatment adherence is critical; non-compliance causes relapse."
Q5: What is the neurological worsening paradox?
"Up to 50% of patients with neurological WD experience paradoxical worsening of neurological symptoms during the first 3-6 months of chelation therapy, particularly with D-penicillamine. The mechanism is thought to be rapid mobilization of hepatic copper stores, causing a transient increase in free serum copper that deposits in the brain. Clinically, patients develop worsening tremor, dystonia, dysarthria, or gait instability. This is NOT treatment failure. Management includes: reassuring the patient that it's usually transient, continuing treatment (discontinuation worsens outcomes), considering dose reduction or slow escalation, switching to trientine (lower risk), and adding zinc acetate. Prevention strategies include starting with low chelator doses and gradual escalation, or using trientine as first-line in neurological patients."
Q6: What are Kayser-Fleischer rings and how are they detected?
"Kayser-Fleischer (KF) rings are pathognomonic for WD and represent copper deposition in Descemet's membrane at the corneal periphery. They appear as golden-brown or greenish discolouration, typically starting superiorly, then inferiorly, eventually forming a complete ring. KF rings are detected by slit-lamp examination by an ophthalmologist—they are not reliably visible to the naked eye, especially in early stages or in lightly pigmented irides. Prevalence varies by phenotype: present in 95% of neurological WD but only 50-60% of purely hepatic WD, and 20-30% of presymptomatic cases. Importantly, absence of KF rings does NOT exclude WD, especially in hepatic presentations. KF rings may fade or disappear with successful chelation therapy."
Q7: How do you screen first-degree relatives?
"All first-degree relatives (siblings, children) of a confirmed WD patient should undergo screening, as siblings have a 25% recurrence risk. The screening protocol includes: (1) serum ceruloplasmin—low in 90% of affected individuals; (2) 24-hour urinary copper—elevated in affected; (3) slit-lamp examination—to detect KF rings; (4) liver function tests (ALT, AST, bilirubin); and (5) ATP7B genetic testing—especially useful if the index case's mutations are known, allowing direct genotyping. If all tests are normal, the individual is unlikely to be affected, but repeat testing in 1-2 years is advised, especially in children. Early detection of presymptomatic individuals allows initiation of treatment before irreversible damage occurs, dramatically improving outcomes."
Q8: When is liver transplantation indicated?
"Liver transplantation is indicated in two main scenarios: (1) Fulminant hepatic failure (Wilsonian crisis)—acute liver failure with Coombs-negative haemolysis, high AST/ALT ratio, low alkaline phosphatase. This is a medical emergency; chelation alone is inadequate. Transplant assessment should be immediate, and these patients are listed as highest priority. Prognostic scores like the New Wilson Index (Nazer score ≥11) predict death without transplant. (2) Decompensated cirrhosis—refractory ascites, variceal bleeding, hepatorenal syndrome, or progressive liver failure despite 3-6 months of chelation therapy. Transplantation is curative for the metabolic defect (donor liver has normal ATP7B), with 5-year survival of 80-90%. Neurological symptoms may also improve post-transplant, though more slowly than hepatic features. Lifelong immunosuppression is required."
Clinical Examination Stations
Long Case Scenario: Young adult with movement disorder and liver disease
Presenting Complaint: 24-year-old male with 18-month history of progressive tremor, slurred speech, and recent jaundice.
Examination Findings:
- General: Jaundiced, cachectic
- Hands: Coarse tremor at rest and with posture; "wing-beating" tremor with arms outstretched
- Face: Mask-like facies, drooling, dysarthria
- Eyes: Slit-lamp reveals golden-brown KF rings bilaterally
- Abdomen: Hepatosplenomegaly, mild ascites
- Neurology: Cogwheel rigidity, bradykinesia, wide-based gait
Key Points to Mention:
- Combination of neurological (movement disorder) and hepatic (cirrhosis) features in young adult strongly suggests Wilson disease
- KF rings are pathognomonic
- Differential diagnosis: Parkinson disease (but too young, no KF rings, no liver disease), Huntington disease (chorea predominant, no liver disease)
- Investigations: Ceruloplasmin, 24h urinary copper, liver copper, ATP7B genetics, MRI brain
- Management: Chelation (trientine preferred given neurological features), zinc, monitor for neurological worsening, consider transplant if decompensated
- Screen first-degree relatives
Common Exam Mistakes
- ❌ Assuming normal ceruloplasmin excludes WD (5-10% have normal levels)
- ❌ Assuming absence of KF rings excludes WD (only 50% of hepatic WD have KF rings)
- ❌ Discontinuing treatment when neurological symptoms worsen initially (neurological worsening paradox is expected)
- ❌ Not screening first-degree relatives
- ❌ Not considering WD in young patient with unexplained liver disease, movement disorder, or psychiatric symptoms
- ❌ Forgetting to mention lifelong treatment requirement
- ❌ Not recognizing fulminant WD as a transplant emergency
13. Patient Explanation (Layperson Language)
What is Wilson Disease?
"Wilson disease is a rare inherited condition where your body cannot get rid of copper properly. We all need a tiny amount of copper from our diet, but in Wilson disease, copper builds up over time, mainly in your liver and brain, and can damage these organs."
Why Does It Happen?
"You inherit Wilson disease from your parents. It's caused by a change (mutation) in a gene called ATP7B. This gene normally helps your liver get rid of extra copper through bile (a digestive fluid). When the gene doesn't work properly, copper accumulates instead of being removed. Both parents need to carry the gene for you to have Wilson disease—you inherit one faulty gene from each parent."
What Are the Symptoms?
"Symptoms usually start in teenagers or young adults, but can appear in children or older adults. Wilson disease can affect different parts of your body:
- Liver problems: Tiredness, yellowing of the skin or eyes (jaundice), swollen belly, feeling unwell. Some people have no symptoms but blood tests show liver damage.
- Movement and brain problems: Shaking hands (tremor), stiffness, difficulty walking, slurred speech, trouble swallowing, mood changes, depression, memory problems.
- Eyes: Golden-brown rings around the colored part of your eye (called Kayser-Fleischer rings). You can't see these yourself—an eye doctor finds them with a special lamp. They don't affect your vision.
Some people are diagnosed before symptoms start, through family screening."
How Is It Diagnosed?
"Your doctor will do several tests:
- Blood tests: Check ceruloplasmin (a protein that carries copper—it's usually low in Wilson disease) and copper levels.
- Urine test: Measure how much copper your body is getting rid of (it's high in Wilson disease).
- Eye examination: An eye doctor uses a slit-lamp to look for Kayser-Fleischer rings.
- Genetic test: Check for the gene mutation.
- Sometimes a liver biopsy (small sample of liver tissue) or brain scan (MRI) is needed."
How Is It Treated?
"The good news is Wilson disease is treatable. Treatment removes extra copper from your body and prevents new copper from building up. You need to take medication for the rest of your life.
- Chelation tablets (like D-penicillamine or trientine): These bind to copper in your body and help remove it through urine. You take them on an empty stomach every day.
- Zinc tablets: These block your intestines from absorbing copper from food. Often used for long-term treatment or in combination with chelation.
- Diet changes: Avoid foods high in copper (like shellfish, liver, nuts, chocolate, mushrooms).
Important: Never stop your medication without talking to your doctor. Stopping treatment can cause serious relapse and harm to your liver and brain.
Some people experience worsening symptoms (especially tremor or stiffness) when starting treatment—this is temporary and doesn't mean the treatment isn't working. Your doctor will monitor you closely."
What If Treatment Doesn't Work?
"In rare severe cases (like sudden liver failure), a liver transplant may be needed. A transplant cures the copper problem because the new liver works normally."
What Happens If Wilson Disease Is Not Treated?
"Without treatment, Wilson disease gets worse over time and can cause severe liver damage (cirrhosis, liver failure) or severe brain and movement problems. Untreated Wilson disease is life-threatening. With treatment, most people live normal, healthy lives."
What About My Family?
"Wilson disease runs in families. If you have Wilson disease, your brothers, sisters, and children should be tested, even if they feel well. Early detection and treatment can prevent symptoms from ever developing."
Living With Wilson Disease
- Take your medication every day as prescribed
- Attend regular check-ups with your doctor
- Avoid alcohol (it can damage your liver further)
- Eat a balanced diet and avoid high-copper foods
- Tell other doctors and dentists you have Wilson disease
- Carry medical alert information
- If you're planning a pregnancy, talk to your doctor—treatment needs to continue but doses may be adjusted
Remember: Wilson disease is manageable with lifelong treatment. Early diagnosis and adherence to therapy mean you can live a full, normal life.
14. References
-
Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089-2111. doi:10.1002/hep.22261. PMID: 18506894
-
Bandmann O, Weiss KH, Kaler SG. Wilson's disease and other neurological copper disorders. Lancet Neurol. 2015;14(1):103-113. doi:10.1016/S1474-4422(14)70190-5. PMID: 25496898
-
European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol. 2012;56(3):671-685. doi:10.1016/j.jhep.2011.11.007. PMID: 22340672
-
Rodríguez-Castro KI, Hevia-Urrutia FJ, Sturniolo GC. Wilson's disease: A review of what we have learned. World J Hepatol. 2015;7(29):2859-2870. doi:10.4254/wjh.v7.i29.2859. PMID: 26689333
-
Weiss KH, Stremmel W. Clinical considerations for an effective medical therapy in Wilson's disease. Ann N Y Acad Sci. 2014;1315:81-85. doi:10.1111/nyas.12455. PMID: 24820352
-
Coffey AJ, Durkie M, Hague S, et al. A genetic study of Wilson's disease in the United Kingdom. Brain. 2013;136(Pt 5):1476-1487. doi:10.1093/brain/awt035. PMID: 23518715
-
Kenney SM, Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat. 2007;28(12):1171-1177. doi:10.1002/humu.20586. PMID: 17680703
-
Pfeiffer RF. Wilson's Disease. Semin Neurol. 2007;27(2):123-132. doi:10.1055/s-2007-971173. PMID: 17390257
-
Lorincz MT. Neurologic Wilson's disease. Ann N Y Acad Sci. 2010;1184:173-187. doi:10.1111/j.1749-6632.2009.05109.x. PMID: 20146697
-
Korman JD, Volenberg I, Balko J, et al. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology. 2008;48(4):1167-1174. doi:10.1002/hep.22446. PMID: 18798626
-
Nazer H, Ede RJ, Mowat AP, Williams R. Wilson's disease: clinical presentation and use of prognostic index. Gut. 1986;27(11):1377-1381. doi:10.1136/gut.27.11.1377. PMID: 3792921
-
Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23(3):139-142. doi:10.1034/j.1600-0676.2003.00824.x. PMID: 12955875
-
Medici V, Rossaro L, Sturniolo GC. Wilson disease--a practical approach to diagnosis, treatment and follow-up. Dig Liver Dis. 2007;39(7):601-609. doi:10.1016/j.dld.2006.12.095. PMID: 17324633
-
Brewer GJ, Johnson VD, Dick RD, Brunberg JA, Kluin KJ, Fink JK. Treatment of Wilson's disease with zinc. XVII: treatment during pregnancy. Hepatology. 2000;31(2):364-370. doi:10.1002/hep.510310214. PMID: 10655259
-
Walshe JM. Penicillamine, a new oral therapy for Wilson's disease. Am J Med. 1956;21(4):487-495. doi:10.1016/0002-9343(56)90066-3. PMID: 13362281
-
Brewer GJ, Dick RD, Johnson V, Wang Y, Yuzbasiyan-Gurkan V, Kluin K, Fink JK, Aisen A. Treatment of Wilson's disease with zinc: XV long-term follow-up studies. J Lab Clin Med. 1998;132(4):264-278. doi:10.1016/s0022-2143(98)90042-8. PMID: 9794699
-
Litwin T, Gromadzka G, Członkowska A. Gender differences in Wilson's disease. J Neurol Sci. 2012;312(1-2):31-35. doi:10.1016/j.jns.2011.08.028. PMID: 21925683
-
Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet. 2007;369(9559):397-408. doi:10.1016/S0140-6736(07)60196-2. PMID: 17276780
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Copper Metabolism
- Ceruloplasmin
Differentials
Competing diagnoses and look-alikes to compare.
- Autoimmune Hepatitis
- Parkinson Disease
- Hemochromatosis
Consequences
Complications and downstream problems to keep in mind.
- Acute Liver Failure
- Cirrhosis
- Movement Disorders