Paeds · neurology-neurodisability-and-neuromuscular
Epilepsy syndromes by age
Also known as Age-related epilepsy syndromes · ILAE 2022 epilepsy syndromes · Paediatric electroclinical syndromes · Self-limited and developmental epileptic encephalopathies · Idiopathic generalised epilepsies
Fellowship guide to the paediatric epilepsy syndromes organised along the developmental timeline, built on the ILAE 2022 Task Force nosology. The page teaches how a syndrome — the cluster of seizure type, EEG signature, age of onset and developmental context — points to a cause, a prognosis and a single best treatment. It walks the neonatal channelopathies through infantile spasms and Dravet syndrome to the self-limited focal epilepsies, childhood and juvenile absence epilepsies, Lennox-Gastaut syndrome and juvenile myoclonic epilepsy, and stresses syndrome-directed antiseizure medicine selection and the drugs that worsen specific syndromes.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture two children with "seizures." One is a previously well seven-year-old who has a few nocturnal facial twitching episodes and EEG centrotemporal spikes; the other is a six-month-old whose development has stalled and who has brief flexion spasms in clusters on a chaotic hypsarrhythmic EEG. Both have epilepsy, but the word means very different things for each. The syndrome label — self-limited epilepsy with centrotemporal spikes in the first, West syndrome in the second — is what tells you the cause, the prognosis and the drug. That is why paediatric epilepsy is taught and examined as syndromes, not as undifferentiated "fits." [1]
The 2017 ILAE operational framework separates three levels of thought. First, you classify the seizure type — focal onset (with awareness preserved or impaired), generalised onset (motor or non-motor), or unknown onset. Second, you classify the epilepsy type along the same focal, generalised or combined axis. Third, where the pattern fits, you assign an epilepsy syndrome, which adds age of onset, EEG features, comorbidities and expected course. The aetiology — structural, genetic, infectious, metabolic, immune or unknown — sits alongside at every level, because a syndrome and its cause are independent questions. [6] [7]
Seizure type
Epilepsy syndrome
Aetiology
The most useful clinical distinction the 2022 nosology makes is between a self-limited syndrome and a developmental and epileptic encephalopathy (DEE). The word "benign" has been retired because it underplays the real cognitive and behavioural morbidity that even the "good" syndromes can carry; "self-limited" means the epilepsy is expected to remit, but it does not guarantee a perfect outcome. A DEE is the opposite end: here the frequent epileptic activity itself drives developmental regression or stagnation, so that controlling the electroencephalographic burden is part of protecting the brain, not just stopping the visible seizures. Holding that distinction in your head reframes the whole age timeline. [1] [8]
Classification
The syndromes line up along the child's developmental timeline, and that timeline is the skeleton on which this whole topic hangs. The ILAE 2022 Task Force published five position papers that together define the complete list: a methodology paper with the master list, one each for syndromes of neonatal and infant onset, childhood onset, the idiopathic generalised epilepsies, and syndromes with onset at a variable age. Reading them as a timeline — rather than as an alphabetical list — is how a clinician actually reasons at the bedside: you hear the age, and only a handful of syndromes remain plausible. [1] [2]
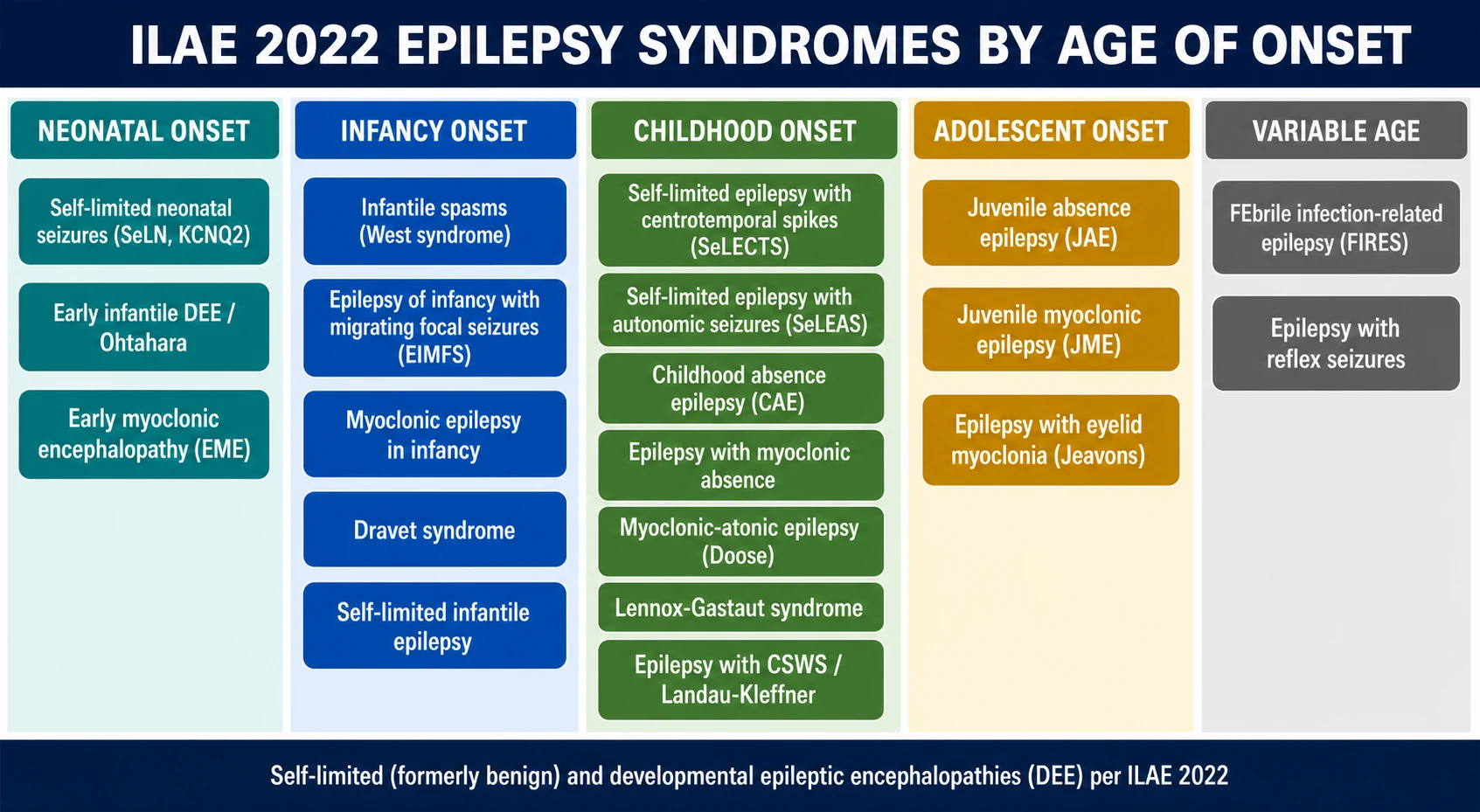
At the neonatal end sit the channelopathies: self-limited neonatal seizures (formerly benign familial neonatal seizures, linked to KCNQ2/3), and the two devastating early encephalopathies — Ohtahara syndrome (early infantile developmental and epileptic encephalopathy with burst-suppression) and early myoclonic encephalopathy. [2] In infancy the list grows to include infantile spasms (West syndrome), epilepsy of infancy with migrating focal seizures, Dravet syndrome, and myoclonic epilepsy in infancy. [2] Childhood brings the self-limited focal epilepsies — self-limited epilepsy with centrotemporal spikes and with autonomic seizures — alongside childhood absence epilepsy, the epilepsy with myoclonic absences, myoclonic-atonic epilepsy (Doose), and Lennox-Gastaut syndrome. [3] The idiopathic generalised epilepsies — childhood absence, juvenile absence and juvenile myoclonic epilepsy, and epilepsy with eyelid myoclonia — span the school-age and adolescent years and are grouped in their own ILAE paper because they share a genetic generalised mechanism. [4]
Epidemiology & Risk Factors
Epilepsy incidence in children is highest at the two extremes of childhood — in the first year of life and again in later childhood — with an overall incidence of roughly 40 to 50 per 100,000 children per year in high-income settings. The severe infant syndromes, although individually rare, account for a disproportionate share of epilepsy-related morbidity, hospital admissions and developmental disability, which is why they dominate the fellowship exam. [1]
The single most important risk factor for the infant DEEs is a monogenic cause, and the family history and fever-sensitivity pattern are the clues that point you toward it. A child with Dravet syndrome usually has de novo pathogenic variants in SCN1A; the broader GEFS+ (genetic epilepsy with febrile seizures plus) spectrum is familial and dominant. Tuberous sclerosis complex is the leading identifiable structural cause of infantile spasms, and its presence changes the first-choice drug to vigabatrin. Perinatal injury, congenital brain malformations and inborn errors of metabolism underlie many of the symptomatic infant encephalopathies. [2]
Pathophysiology
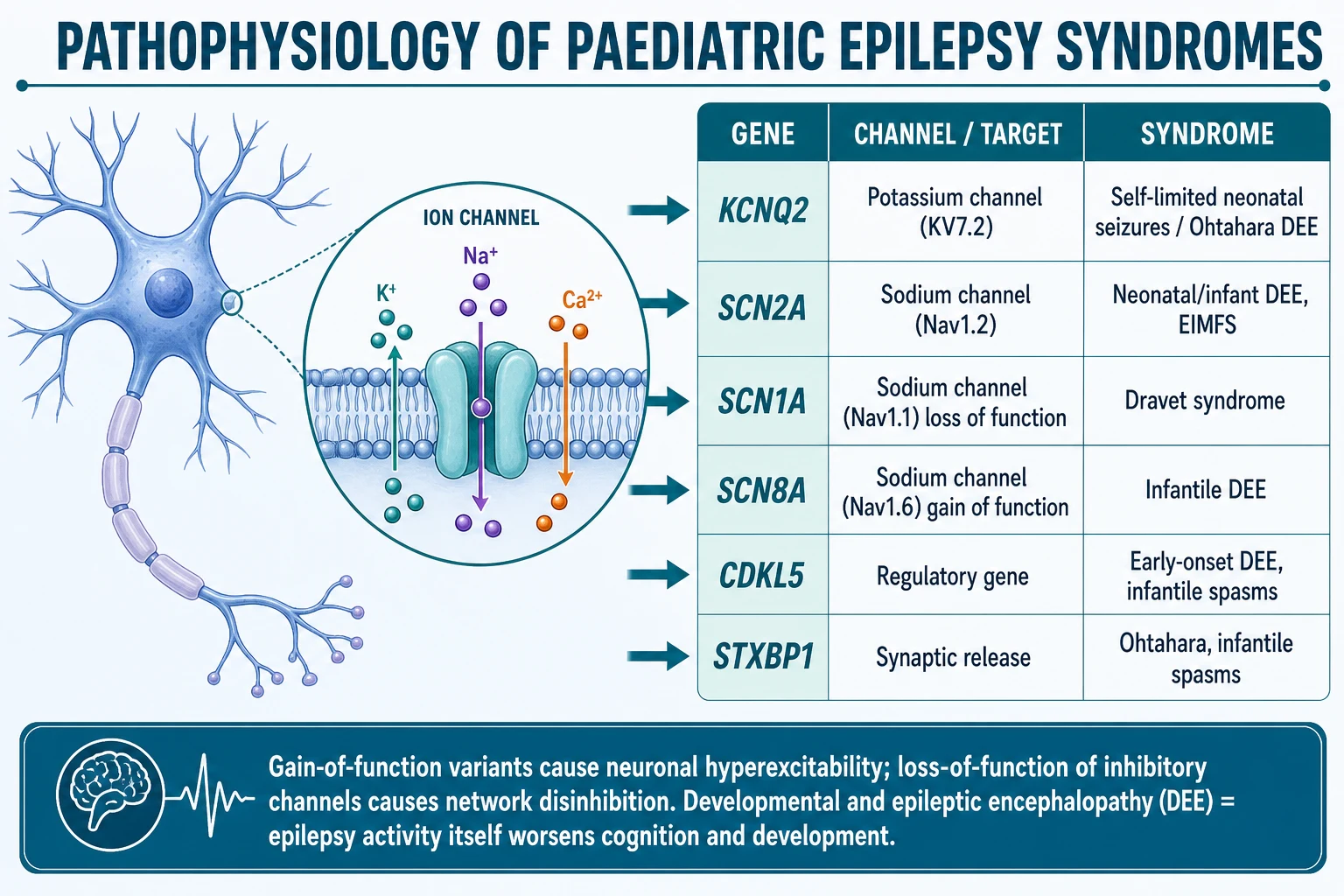
Why should seizures cluster at specific ages? The answer lies in the developing brain and the genes that wire it. Many paediatric epilepsy syndromes are channelopathies — disorders of the voltage-gated or ligand-gated ion channels that control neuronal firing — and the expression of those channels changes dramatically across development. A variant that causes chaotic neonatal seizures may be silent later, while a different sodium-channel variant produces no problem until adolescence. [2]
The sodium-channel story is the one to hold. The distinction between gain-of-function and loss-of-function matters clinically because it predicts both the syndrome and the drug. A gain-of-function variant in SCN2A (the Nav1.2 channel) makes neurons fire too readily and produces neonatal or infantile epilepsy that may respond to sodium-channel blockers. The opposite is true for SCN1A (Nav1.1): here a loss-of-function variant removes inhibition from the circuit, producing Dravet syndrome, and giving a sodium-channel blocker — which further suppresses the remaining inhibitory neurons — makes the child worse. That single mechanism explains the most dangerous prescribing error in paediatric epilepsy. [2] [10]
The generalised epilepsies operate through a different circuit. Childhood and juvenile absence epilepsy and juvenile myoclonic epilepsy are thought to arise from abnormal thalamocortical oscillations that generate the rhythmic generalised spike-wave discharges on EEG. These are genetic generalised epilepsies — polygenic in most cases — and they share a tendency to be provoked by sleep deprivation, hyperventilation and (in photosensitive individuals) flickering light. [4]
Clinical Presentation
The presenting feature is usually a seizure, but the kind of seizure and its context are what name the syndrome. An infantile spasm is a brief, symmetric contraction of axial and limb muscles — a sudden jackknife or "salaam" — that repeats in clusters of twenty or thirty over a few minutes, most often on waking. [2] A Dravet seizure in the first year is a prolonged febrile or afebrile hemiclonic convulsion, often lasting more than fifteen minutes, sometimes alternating sides, and frequently triggered by fever or hot baths. [10] A childhood absence is a brief (five to ten second) staring spell with abrupt onset and offset, with no postictal confusion, occurring many times a day and often visible to a teacher before the parents notice. [4]
JERKS
The self-limited focal epilepsies have distinctive semiologies that examiners reward you for recognising. Self-limited epilepsy with centrotemporal spikes (SeLECTS, formerly benign rolandic epilepsy) presents with nocturnal seizures that begin with sensory disturbance and twitching in one corner of the mouth, sometimes spreading to the arm, with drooling and guttural speech — in an otherwise entirely normal school-age child. Self-limited epilepsy with autonomic seizures (SeLEAS, formerly Panayiotopoulos syndrome) produces autonomic features, especially nausea, retching and vomiting, with pallor and deviation of the eyes, and the seizures are often long and misdiagnosed as encephalitis or migraine. [3]
Differential Diagnosis
The differential of a child with "seizures" collapses quickly once you fix the age and the EEG, but a few non-epileptic mimics catch the unwary. Infantile spasms can be confused with benign myoclonus of infancy, colic, or Sandifer syndrome from gastro-oesophageal reflux; the EEG distinguishes them, because the chaotic hypsarrhythmia of West syndrome is unmistakable. Brief absences can look like daydreaming or focal impaired-awareness seizures; provoking one with hyperventilation and capturing a 3 Hz spike-wave on EEG settles it. [3] [4]
Childhood absence epilepsy
Focal impaired-awareness seizure
Daydreaming / inattention
The trap that examiners probe is the generalised epilepsy treated as focal. A child with juvenile myoclonic epilepsy may first present with a single early-morning convulsion and the myoclonic jerks are missed on history; starting carbamazepine can worsen the myoclonus and the generalised spike-wave. Always ask directly about morning jerks, about absences, and about photosensitivity before prescribing, and request an EEG that includes hyperventilation, photic stimulation and sleep, because the answers change the drug. [4]
Clinical & Bedside Assessment
The focused history is the single most diagnostic tool in paediatric epilepsy. Ask for the age of onset, the exact semiology of each seizure type (a parent video is worth a thousand words), any developmental regression or plateau, fever or illness as a trigger, and a careful family history of seizures, febrile convulsions and learning difficulty. A child whose development was normal until seizures began, then stalled, has a DEE until proven otherwise; a child who is entirely well between seizures and has a normal developmental trajectory points toward a self-limited syndrome. [1] [3]
At the bedside you can actively provoke an absence seizure. Ask the child to hyperventilate for two to three minutes — blowing on a pinwheel or a paper tissue — and watch for a brief pause in the blowing, a stare, and a flutter of the eyelids. A true absence interrupts the task and ends abruptly; if you have an EEG running, the 3 Hz spike-wave appears within seconds. This single manoeuvre is both diagnostic and deeply satisfying to demonstrate, and examiners expect you to know it. [4]
The neurological examination is usually normal in the self-limited and idiopathic generalised epilepsies; a focal deficit, a cutaneous lesion (ash-leaf, shagreen patch, adenoma sebaceum of tuberous sclerosis) or microcephaly should redirect you toward a structural or syndromic cause. Skin examination with a Wood's lamp is part of the work-up of any infant with spasms, because tuberous sclerosis changes both the prognosis and the first-choice drug. [2]
Investigations
The EEG is the core investigation, and in many syndromes it is near-pathognomonic. The patterns worth memorising are hypsarrhythmia (chaotic, high-voltage, asynchronous slow waves with multifocal spikes) in West syndrome, the 3 Hz generalised spike-wave of childhood absence epilepsy, the centrotemporal spikes activated in sleep of SeLECTS, the slow spike-wave at 1.5 to 2.5 Hz of Lennox-Gastaut syndrome, the electrical status epilepticus in slow sleep of the CSWS and Landau-Kleffner syndromes, and the photo-paroxysmal response of juvenile myoclonic epilepsy and epilepsy with eyelid myoclonia. A sleep-deprived EEG with activation procedures is more sensitive than a routine awake record. [3] [4]
Neuroimaging is not universal. In a child with a classic self-limited syndrome (SeLECTS, childhood absence) and a typical EEG, an MRI is often unnecessary, because the clinical-electrical pattern is so characteristic. MRI is essential, however, in any focal epilepsy that is not a recognised self-limited syndrome, in any infant with spasms of unknown cause, and whenever the seizure semiology, the examination or the developmental trajectory suggests a structural lesion. [7]
Genetic testing has moved from a research tool to a first-line investigation in the infant DEEs. An early gene panel or, increasingly, whole-exome sequencing identifies a monogenic cause in a substantial proportion of children with infantile spasms, Dravet syndrome and the early-onset channelopathies; the result changes counselling, prognosis and sometimes treatment. Metabolic studies (plasma amino acids, urine organic acids, lactate, ammonia) and, where appropriate, autoimmune encephalitis antibody panels close the differential in atypical or deteriorating cases. [2] [10]
Management — Resuscitation
Most epilepsy syndrome presentations are not emergencies, but two scenarios are. The first is a prolonged convulsive seizure, most often a Dravet-type prolonged febrile seizure. The standard paediatric status epilepticus ladder applies: a first-line benzodiazepine — lorazepam 0.1 mg per kg intravenously, or buccal midazolam 0.15 mg per kg or intranasal midazolam when no intravenous access exists — followed by a second-line agent such as levetiracetam or phenytoin if the seizure continues, and escalation to intensive care for refractory status. The key syndrome-specific point is that a child with known or suspected Dravet syndrome needs a written emergency plan, because their seizures are prolonged, fever-triggered and recurrent. [10]
Emergency management of a prolonged convulsive seizure in a child with a known or suspected DEE
Assess airway, breathing, circulation; give high-flow oxygen; check glucose and temperature
First-line benzodiazepine: lorazepam 0.1 mg/kg IV, or buccal midazolam 0.15 mg/kg if no IV access (repeat once after 5 to 10 minutes if still seizing)
Second-line if ongoing at 5 to 10 minutes: levetiracetam 40 to 60 mg/kg IV OR phenytoin 20 mg/kg IV (avoid phenytoin if Dravet suspected)
Refractory status beyond 30 minutes: intensive care, continuous infusion (midazolam, propofol or thiopent), intubation and EEG monitoring
Treat the trigger (fever, infection); for Dravet, review the maintenance regimen and the medicines to avoid
The second emergency is a cluster of epileptic spasms or non-convulsive status in an infant; the urgency is developmental, not just seizural, because ongoing epileptic encephalopathy erodes development. Here the immediate step is diagnostic — a prolonged video-EEG to confirm the spasms and the hypsarrhythmia — followed by rapid initiation of syndrome-specific treatment rather than generic antiseizure cover. [2] [9]
Management — Definitive & Stepwise
Once the syndrome is named, treatment is syndrome-directed: you choose the single best drug for that syndrome, not a generic antiseizure medicine. The principle rests on the observation that the right first drug controls seizures far more often than a wrong one, and that a wrong first drug can worsen the syndrome — the most examinable point in this topic. [1]
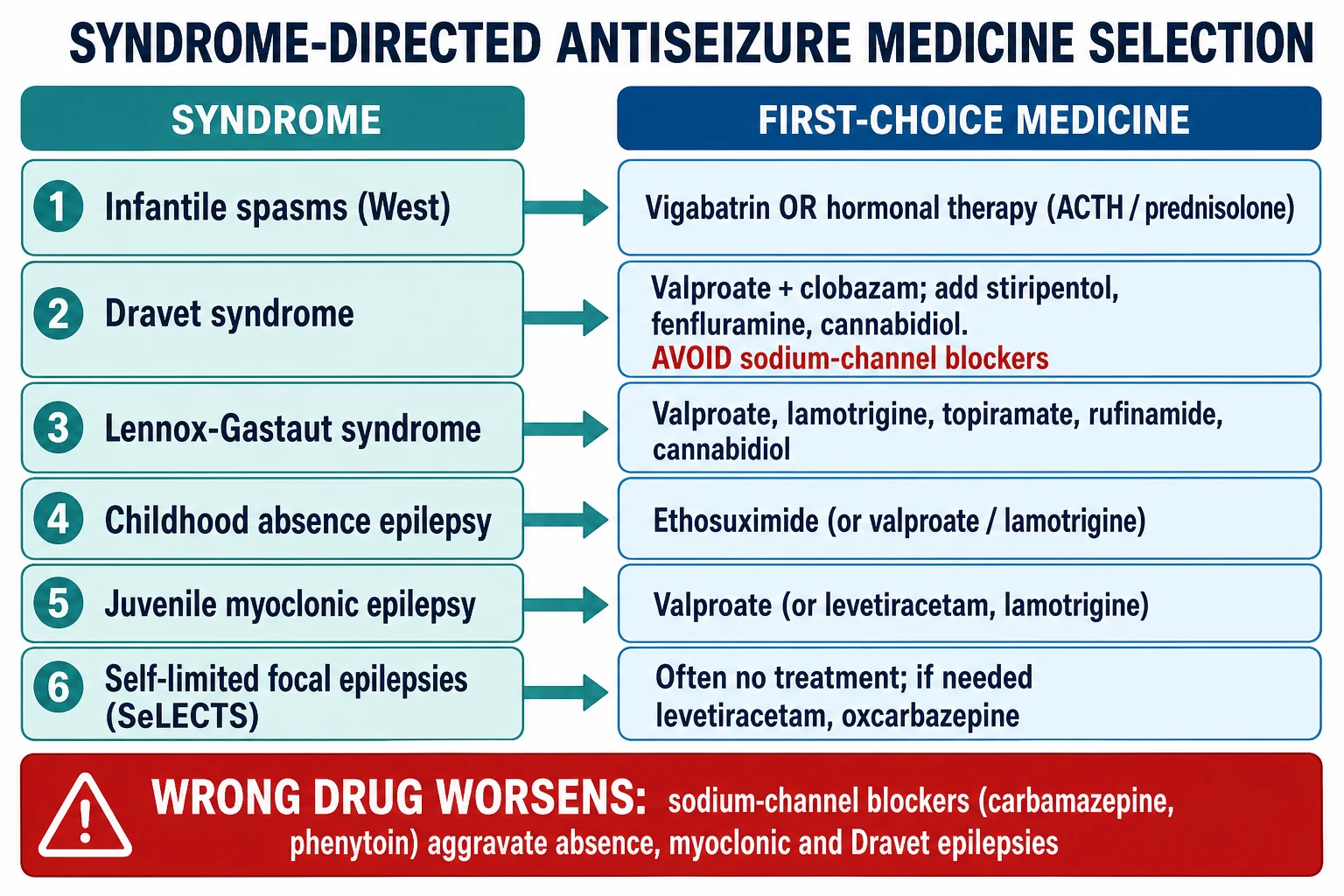
For infantile spasms (West syndrome), first-line treatment is vigabatrin or hormonal therapy (adrenocorticotrophic hormone, or high-dose prednisolone). Vigabatrin is preferred when the cause is tuberous sclerosis, and hormonal therapy (ACTH or oral prednisolone) is favoured for other causes; the Cochrane review found hormonal therapy more effective than vigabatrin for short-term spasm cessation in non-tuberous-sclerosis cases, though vigabatrin carries a risk of irreversible visual field loss with prolonged use. Treatment must start early, because time-to-treatment is a major determinant of developmental outcome. [9]
For Dravet syndrome, the foundation is sodium valproate combined with a benzodiazepine such as clobazam, with stiripentol, fenfluramine and cannabidiol added for refractory disease. The non-negotiable rule is to avoid sodium-channel blockers — carbamazepine, oxcarbazepine, lamotrigine and phenytoin — because they worsen the seizures by further suppressing the already-deficient inhibitory neurons. A ketogenic diet is an effective adjunct. [10]
Sodium valproate (syndrome: Dravet, JME, LGS, JAE)
Dose
Maintenance 20 to 30 mg/kg/day in two divided doses
For the generalised epilepsies, ethosuximide is first-line for childhood absence epilepsy where there are no generalised tonic-clonic seizures (valproate is preferred when generalised tonic-clonic seizures coexist, because ethosuximide does not cover them). Valproate remains first-line for juvenile myoclonic epilepsy and juvenile absence epilepsy because of its broad-spectrum cover across absence, myoclonic and tonic-clonic seizures; where valproate is unsuitable (teratogenicity in a young woman), levetiracetam or lamotrigine are alternatives, though lamotrigine may worsen myoclonus. [4]
For Lennox-Gastaut syndrome, treatment is broad-spectrum and often only partially effective: valproate, lamotrigine, topiramate, rufinamide and cannabidiol each have evidence, and the practical aim is seizure reduction and protection from drop attacks rather than complete freedom from seizures. For the self-limited focal epilepsies, many children need no treatment at all because the seizures are infrequent and nocturnal; when treatment is required, levetiracetam or oxcarbazepine are effective and the epilepsy remits by adolescence. [3]
Specific Subtypes & Scenarios
West syndrome is the archetype of a treatable DEE. The triad is epileptic spasms, hypsarrhythmia on EEG, and psychomotor regression or developmental delay, with peak onset between four and eight months. The parents describe brief symmetric jerks — flexion or extension of the neck, trunk and limbs — that cluster on waking, often dozens at a time, and they will often have noticed that their baby has stopped smiling, reaching or rolling. The EEG shows the chaotic, high-voltage hypsarrhythmia, and treatment with vigabatrin or hormonal therapy aims not only to stop the spasms but to resolve the electrical encephalopathy and protect development. Aetiology drives outcome: cryptogenic cases do better than those with an identified structural or genetic cause, and tuberous sclerosis, hypoxic-ischaemic injury and chromosomal abnormalities are the commonest identified causes. [2] [9]
Dravet syndrome begins in the first year of life with prolonged febrile or afebrile hemiclonic seizures, often lasting more than fifteen minutes and sometimes alternating sides, triggered by fever, hot environments or vaccination. Over the next two years other seizure types emerge — myoclonic, atypical absence, focal and absence — and development, initially normal, slows and regresses with ataxia and intellectual disability. Around seventy percent carry a pathogenic loss-of-function variant in SCN1A. The syndrome-specific management rule — avoid sodium-channel blockers, build on valproate and a benzodiazepine, add stiripentol, fenfluramine or cannabidiol — and a written emergency plan for prolonged seizures are the two things that change the course of the disease. [10]
West syndrome
Dravet syndrome
Lennox-Gastaut
Lennox-Gastaut syndrome is the refractory DEE of early childhood. The triad is multiple seizure types — tonic, atonic and atypical absence most characteristically, with drop attacks that cause injury — an interictal EEG of slow (1.5 to 2.5 Hz) generalised spike-wave, and cognitive impairment that is usually present at onset and often progresses. Onset is between one and eight years, with a peak around three to five. Treatment is broad-spectrum and the realistic goal is reduction rather than remission; a rescue plan for non-convulsive status and protective headgear for drop attacks are part of holistic care. [3]
Juvenile myoclonic epilepsy sits at the other end of the prognosis spectrum. It begins in adolescence with early-morning myoclonic jerks — the teenager who drops or flings a toothbrush or a mug at breakfast — often followed by a generalised tonic-clonic seizure, and the EEG shows fast (4 to 6 Hz) generalised polyspike-wave with a photo-paroxysmal response in many. Valproate is first-line and highly effective, and the long-standing debate is whether treatment is lifelong; most authorities now regard it as a chronic epilepsy requiring long-term medication, because withdrawal carries a high relapse rate, and the key lifestyle measures are protecting sleep and avoiding alcohol excess. [4]
Complications & Pitfalls
The gravest pitfall is prescribing a sodium-channel blocker to the wrong syndrome. Carbamazepine, oxcarbazepine, lamotrigine and phenytoin worsen the absence, myoclonic and spike-wave burden of the genetic generalised epilepsies and the seizures of Dravet syndrome; a child whose epilepsy "escalates" after starting a new antiseizure medicine should have the diagnosis — not just the dose — re-examined. [4] [10]
The developmental cost of untreated DEEs is the second major theme. In West syndrome, time-to-treatment correlates with developmental outcome, so delays in recognising infantile spasms or in obtaining an EEG translate directly into lost cognition. In the CSWS and Landau-Kleffner syndromes, the continuous spike-wave of slow sleep erodes language and behaviour even when the visible seizures are few, so the EEG — not the seizure count — guides treatment. [3]
Sudden unexpected death in epilepsy (SUDEP) is a real risk in the severe childhood epilepsies, concentrated in children with frequent generalised tonic-clonic seizures, nocturnal seizures and structural brain disease. Counselling families on SUDEP — when, how and whom to counsel — is a fellowship-level communication skill: the guidance is to discuss it with families of children at higher risk, to advise nocturnal supervision or seizure alarms where appropriate, and to pursue seizure freedom aggressively, because seizure control is the most effective SUDEP prevention. [1]
Prognosis & Disposition
The prognosis separates sharply along the self-limited versus DEE line. The self-limited focal epilepsies and the idiopathic generalised epilepsies of childhood and adolescence remit or respond to a single drug in the great majority; SeLECTS and SeLEAS remit by mid-adolescence even untreated, and the main task is to avoid over-investigation and over-treatment. [3] Childhood absence epilepsy remits in roughly two-thirds by adolescence, though a minority evolves into juvenile absence or juvenile myoclonic epilepsy. [4]
At the DEE end, the prognosis is determined by the aetiology and the response to treatment. West syndrome outcome ranges from normal cognition in a minority of cryptogenic cases that are treated early, to severe intellectual disability and refractory epilepsy in those with an identified structural or genetic cause. [9] Dravet syndrome is lifelong, with intellectual disability and gait disorder in most, though the newer therapies have reduced seizure frequency and improved quality of life. [10] Juvenile myoclonic epilepsy, by contrast, is typically a well-controlled lifelong epilepsy in which the question is not cure but adherence and lifestyle. [4]
Disposition in practice means a named syndrome, a single best drug, a written emergency plan for prolonged seizures, and a clear follow-up pathway — shared care between general paediatrics and a neurology service for the complex epilepsies, and primary or general paediatric follow-up for the self-limited syndromes. Transition to adult epilepsy care is planned early in adolescence for the chronic epilepsies, because the handover of a young person with a DEE or juvenile myoclonic epilepsy is itself a clinical event that needs preparation. [1]
Special Populations
The child with a developmental and epileptic encephalopathy and complex needs embodies the multidisciplinary reality of severe paediatric epilepsy: medication regimens that interact with ketogenic diets, rescue medicines that school staff must administer, feeding and communication needs, and a family carrying enormous psychological and logistical weight. Coordinating that care — neurology, developmental paediatrics, dietetics, psychology, school and respite — is the general paediatrician's role as much as the neurologist's. [1]
The child with a sodium-channelopathy (Dravet and related SCN syndromes) needs a MedicAlert-style alert against sodium-channel blockers, which may be given inadvertently during a febrile illness or in perioperative care, and a plan that covers common triggers: fever, hot environments, over-heating, intercurrent infection and (in some) specific vaccinations. The family must know which drugs to refuse. [10]
The adolescent with juvenile myoclonic epilepsy is the population where lifestyle dominates: sleep deprivation, alcohol excess and strobe exposure reliably trigger seizures, and teratogenicity counselling around valproate is a defining conversation for a young woman, because the syndrome is lifelong and pregnancy planning must begin years in advance. [4]
In Australia and Aotearoa New Zealand, epilepsy syndrome care is delivered through general paediatricians in regional centres and paediatric neurology services in the major children's hospitals, with the Royal Children's Hospital Melbourne and similar guidelines underpinning first-line drug choice. Equity of access to EEG, genetic testing and subspecialist care is a real challenge for Indigenous and remote children, and telehealth-supported shared care is the working model. [1]
Evidence, Guidelines & Regional Differences
The ILAE 2022 Task Force papers are the current global standard for syndrome definition. The five papers — the methodology and master list (Wirrell), neonatal and infant onset (Zuberi), childhood onset (Specchio), the idiopathic generalised epilepsies (Hirsch), and variable-age onset (Riney) — together define each syndrome's mandatory and exclusionary criteria and are the reference point for every guideline that follows. [1] [2] [3] [4] [5]
The Hancock Cochrane review of infantile spasms treatment underpins the hormonal-versus-vigabatrin decision: hormonal therapy (ACTH or oral prednisolone) was more effective than vigabatrin for short-term resolution of spasms in non-tuberous-sclerosis cases, while vigabatrin is preferred for tuberous sclerosis and carries the risk of visual field constriction. [9] The Cross review of Dravet syndrome summarises the evidence for stiripentol, fenfluramine and cannabidiol, which have transformed the management of what was previously a uniformly refractory epilepsy. [10]
The regional differences are modest in principle — all guidelines now follow the ILAE nosology and stress syndrome-directed treatment — but the drug availability and reimbursement differ. The UK NICE guideline on epilepsies in children, young people and adults recommends syndrome-directed first-line therapy and flags the drugs that worsen specific syndromes. [4] Fenfluramine and cannabidiol are funded in some jurisdictions and not others, which affects real-world Dravet and Lennox-Gastaut management; the general principle — broad-spectrum, syndrome-matched, avoiding sodium-channel blockers — is universal. [10]
Exam Pearls
A handful of one-liners carry disproportionate marks. West syndrome is the triad of epileptic spasms, hypsarrhythmia and developmental regression, peaking at four to eight months; treat early. Dravet syndrome is prolonged febrile hemiclonic seizures in the first year, SCN1A, and avoid sodium-channel blockers. Childhood absence epilepsy is 3 Hz spike-wave provoked by hyperventilation, first-line ethosuximide. Juvenile myoclonic epilepsy is early-morning myoclonic jerks with a photo-paroxysmal EEG response, first-line valproate. SeLECTS is nocturnal orofacial sensory-motor seizures with centrotemporal spikes in an otherwise normal child, often needing no treatment. [2] [3] [4]
Hancock Cochrane review — infantile spasms (2013)
Population: Infants with epileptic spasms (West syndrome)
Key finding
Hormonal therapy led to earlier resolution of spasms than vigabatrin in non-tuberous-sclerosis cases; vigabatrin carried a risk of visual field loss
Practice change
Hormonal therapy is first-line for non-tuberous-sclerosis infantile spasms; vigabatrin is preferred for tuberous sclerosis; treat early for best developmental outcome
Finally, remember that "benign" is retired. The self-limited syndromes are expected to remit, but they are not always cognitively benign — SeLECTS can carry subtle attention and learning difficulties, and reading the EEG correctly (and not over-treating) is part of good care. Conversely, a DEE is never benign, and the developmental cost of delay is the reason a suspicious infant EEG is an urgent, not a routine, investigation. [1] [8]
References
- [1]Wirrell EC, Scheffer IE, Berkovic S, et al. Methodology for classification and definition of epilepsy syndromes with list of syndromes: Report of the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503715
- [2]Zuberi SM, Wirrell E, Yozawitz E, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503712
- [3]Specchio N, Wirrell EC, Scheffer IE, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503717
- [4]Hirsch E, French J, Scheffer IE, et al. ILAE definition of the Idiopathic Generalized Epilepsy Syndromes: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503716
- [5]Riney K, Bogacz A, Somerville E, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503725
- [6]Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia, 2017.PMID 28276060
- [7]Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia, 2017.PMID 28276062
- [8]Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia, 2010.PMID 20196795
- [9]Hancock EC, Osborne JP, Edwards SW. Treatment of infantile spasms. Cochrane Database Syst Rev, 2013.PMID 23740534
- [10]Cross JH, Caraballo RH, Nabbout R, Vigevano F, Guerrini R, Lagae L. Dravet syndrome: Treatment options and management of prolonged seizures. Epilepsia, 2019.PMID 31904119