Paeds · respiratory-sleep-and-airway
Cystic fibrosis: diagnosis and screening
Also known as CF diagnosis · Newborn bloodspot screening for cystic fibrosis · Sweat chloride test · CFTR mutation testing · Mucoviscidosis
Fellowship guide to diagnosing and screening for cystic fibrosis in children: recognising the child whose salty skin, chronic wet cough, greasy stools and poor growth should trigger a sweat test; how newborn bloodspot screening with immunoreactive trypsinogen and CFTR mutation panels finds most infants before symptoms; confirming the diagnosis with the sweat chloride test and genetic analysis using the Cystic Fibrosis Foundation and ECFS criteria; interpreting an intermediate sweat chloride and the CRMS/CFSPID equivocal-screen category; the differential from other causes of failure to thrive and chronic suppurative lung disease; the meconium ileus, pseudo-Bartter and false-negative-screen traps; and the ANZ, UK and North American differences in screening protocols.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the six-week-old whose parents mention that she tastes salty when they kiss her, has passed greasy, foul stools since birth, and has barely gained weight despite feeding well, with a rattly cough that has never quite cleared. That combination — salt, malabsorption and a wet chest in a baby who is not thriving — is the clinical shorthand for cystic fibrosis, and it should send your hand straight to a sweat test. [5] [1]
Cystic fibrosis is an autosomal recessive multisystem disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator, the CFTR gene on chromosome seven. CFTR is a chloride channel at the surface of epithelial cells, and when it fails, secretions across the airways, pancreas, gut, sweat glands and reproductive tract become thick and dehydrated. The disease is named for the fibrotic, cyst-like change once seen in the pancreas. [6] [5]
Why this matters at fellowship level is that diagnosis is time-critical. Finding cystic fibrosis before irreversible lung damage or malnutrition sets in changes the child's whole trajectory, so the examinable skill is knowing both how the screening pathway works and when to reach for a sweat test yourself in a child whose screen was negative or was never done. [10] [1]
Classification
The most useful way to frame a suspected case is to answer three linked questions: does this child have CFTR dysfunction, which mutations do they carry, and what does that predict for the pancreas and lungs. Diagnosis, genotype and phenotype travel together, and each shapes the next step. [1] [6]
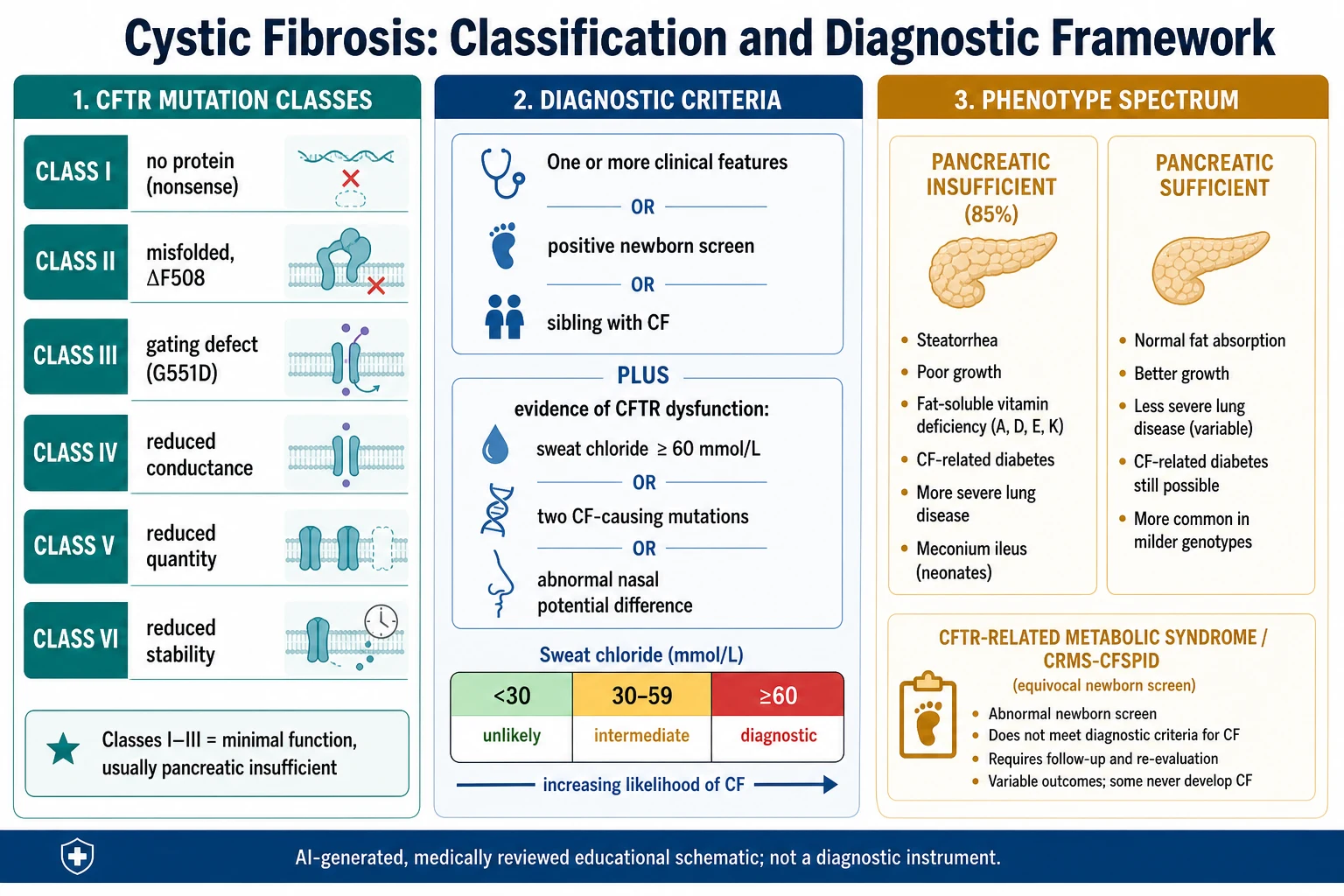
The diagnostic criteria are the first axis. A diagnosis of cystic fibrosis requires one clinical feature, a positive newborn screen, or a sibling with CF, plus objective evidence of CFTR dysfunction. That evidence is a sweat chloride at or above sixty millimoles per litre, two CF-causing mutations, or an abnormal nasal potential difference. A sweat chloride under thirty makes CF unlikely, and thirty to fifty-nine is intermediate. [1] [3]
The second axis is the CFTR genotype, grouped into six mutation classes. Classes one to three, including the common ΔF508 (Phe508del), leave little or no working channel and usually cause pancreatic insufficiency. Classes four to six leave some residual function and tend toward milder, pancreatic-sufficient disease. Genotype guides both prognosis and, increasingly, mutation-specific modulator therapy. [6] [5]
The numbers that anchor your viva
Epidemiology & Risk Factors
Cystic fibrosis is the commonest life-limiting autosomal recessive disorder in people of European ancestry, affecting roughly one in two to three thousand live births, with a carrier frequency of about one in twenty-five. It is less common but by no means absent in other populations, which matters because screening panels built around European mutations miss more cases in non-European children. [5] [6]
The single risk factor that defines the disease is inheritance: a child develops cystic fibrosis only by inheriting a CF-causing mutation from each carrier parent. Two carrier parents have a one-in-four risk with each pregnancy, so a family history of CF, of a sibling with the condition, or of consanguinity raises the pre-test probability substantially. [6] [1]
Because inheritance is fixed at conception, the modifiable questions are about detection rather than prevention. Universal newborn screening, carrier screening in couples planning a family, and access to specialist CF care determine how early and how well a child is diagnosed and treated. Populations with less complete screening, or with mutations poorly covered by local panels, are diagnosed later and often only once symptoms appear. [7] [12]
Pathophysiology
The teaching model runs from one broken channel to disease in every exocrine organ, and it turns on a single idea: without working CFTR, epithelial surfaces cannot move chloride and water properly, so their secretions dehydrate and thicken. That one change explains the salty sweat, the blocked pancreatic ducts and the sticky airway mucus. [6] [5]
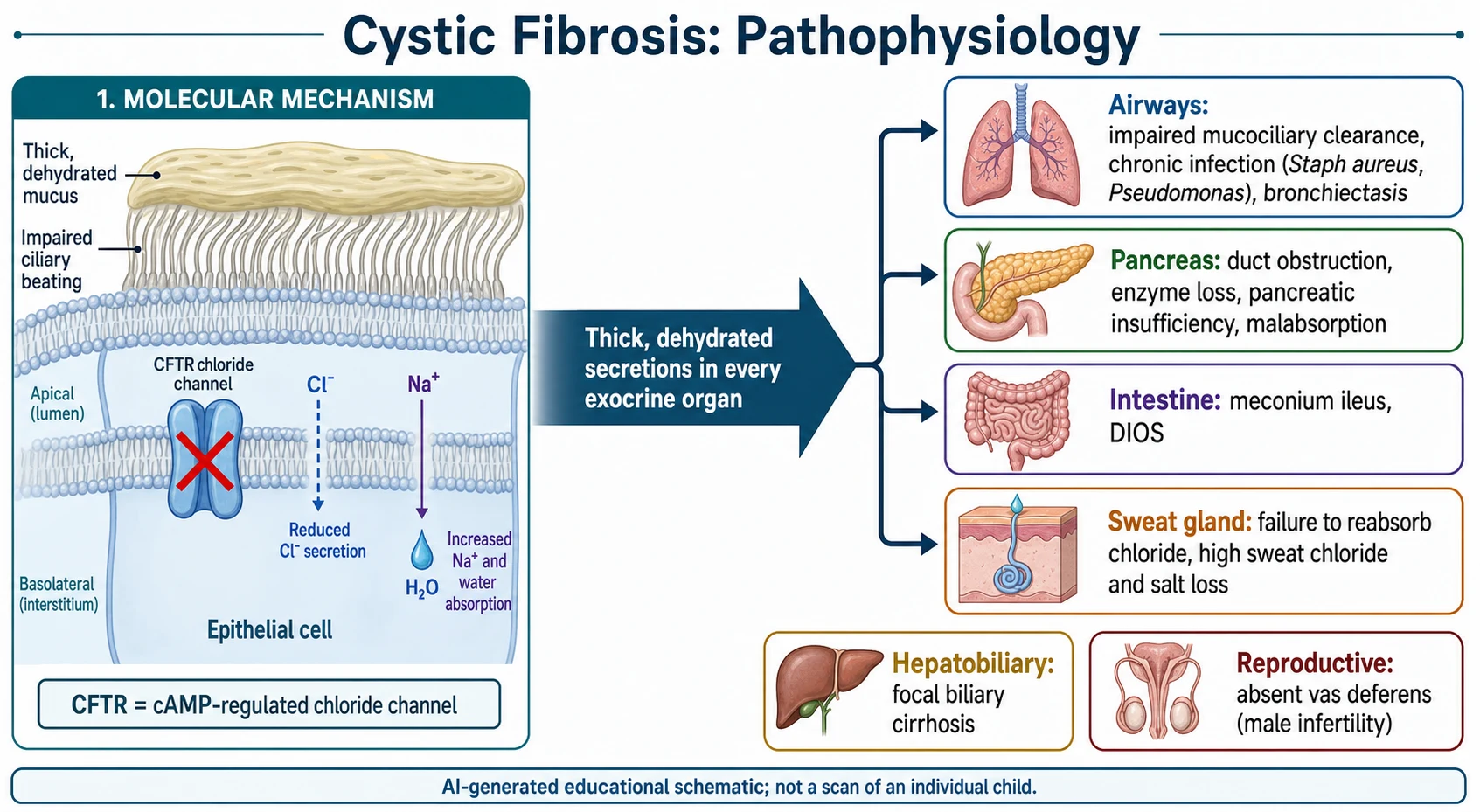
CFTR normally sits at the apical membrane and, when activated, secretes chloride into the lumen while regulating sodium and water movement. In most epithelia the defect leaves the airway surface liquid dehydrated, so mucus becomes viscous and mucociliary clearance fails. Stagnant mucus is then colonised, classically by Staphylococcus aureus early and Pseudomonas aeruginosa later, driving the chronic infection and inflammation that scar the lungs. [5] [6]
The same dehydration damages other organs. In the pancreas, thick secretions plug the ducts, enzymes are lost and fat and protein are malabsorbed, causing steatorrhoea and poor growth; in utero this can present as meconium ileus. Salt loss through sweat can cause hyponatraemic, hypochloraemic dehydration, and thickened bile and genital secretions explain later hepatobiliary disease and male infertility from absent vas deferens. [6] [5]
Clinical Presentation
The presentation now comes in two very different ways. The commonest is the well-looking, asymptomatic newborn flagged by a positive bloodspot screen, whose diagnosis is confirmed before any organ damage. The second is the child who presents clinically because they were never screened, screened negative, or come from a region without screening. Recognising the second child is where marks are won. [9] [3]
The classic clinical picture in infancy is failure to thrive with a chronic wet cough and greasy, bulky, offensive stools. Parents may report that the baby tastes salty. In the newborn period, cystic fibrosis can present dramatically as meconium ileus with bilious vomiting, abdominal distension and failure to pass meconium, which is CF until proven otherwise. [5] [1]
| Age | Typical presentation | Action |
|---|---|---|
| Antenatal / newborn | Echogenic bowel on scan, or meconium ileus | Sweat test and genetics; do not wait for the screen alone |
| Infant | Positive newborn screen, or failure to thrive with wet cough and steatorrhoea | Refer to CF centre for confirmatory sweat test |
| Toddler / child | Recurrent chest infection, rectal prolapse, pseudo-Bartter, salty skin | Sweat test even if the newborn screen was negative |
| Older child / adolescent | Bronchiectasis, nasal polyps, pancreatitis, or male infertility | Consider non-classic CF; sweat test and extended genetics |
Beyond infancy the picture broadens. Older children may present with recurrent or persistent pneumonia, established bronchiectasis, nasal polyps, recurrent pancreatitis, rectal prolapse, or pseudo-Bartter syndrome with hypochloraemic metabolic alkalosis after salt loss in hot weather. Milder, pancreatic-sufficient genotypes may not declare themselves until later childhood or even adulthood. [5] [3]
Differential Diagnosis
Sort the differential by the presenting syndrome rather than trying to hold the whole disease in mind at once. The three doorways into a CF diagnosis are failure to thrive with malabsorption, chronic or recurrent suppurative lung disease, and salt-loss dehydration — each has its own mimics that a sweat test and genetics resolve. The aim is to keep CF on the list whenever these patterns recur. [3] [5]
Cystic fibrosis
the unifying answer
- Salty sweat, chronic wet cough
- Steatorrhoea and failure to thrive
- High sweat chloride, CFTR mutations
- Refer to a CF centre
Other malabsorption
- Coeliac disease, food allergy
- Shwachman-Diamond syndrome
- Normal sweat chloride
- Coeliac serology, marrow, genetics
Other suppurative lung disease
- Primary ciliary dyskinesia
- Immunodeficiency, aspiration
- Normal sweat chloride
- Ciliary studies, immune workup
Other salt loss
- Bartter and Gitelman syndromes
- Adrenal insufficiency
- Normal sweat chloride
- Renal and endocrine workup
For failure to thrive with malabsorption, the main alternatives are coeliac disease, cow's-milk protein allergy and the rarer Shwachman-Diamond syndrome, which shares pancreatic insufficiency but has a normal sweat chloride and marrow failure. For chronic suppurative lung disease, primary ciliary dyskinesia, immunodeficiency and recurrent aspiration all mimic CF, and a normal sweat chloride helps redirect the workup. [3] [5]
For salt-loss dehydration, pseudo-Bartter from CF looks biochemically like the renal tubulopathies Bartter and Gitelman syndrome, and can mimic adrenal salt-wasting. The distinguishing move is simple: a raised sweat chloride points to CF, whereas the renal tubulopathies have a normal sweat chloride with characteristic urinary electrolyte patterns. Whenever these syndromes recur or cluster, do the sweat test. [1] [3]
Clinical & Bedside Assessment
Assessment starts with a targeted history and growth review. Plot weight, length and head circumference on the growth chart, ask about stool pattern and greasiness, the character and duration of any cough, salt cravings or salty skin, and the newborn screening result. Take a full family history for CF, affected siblings, unexplained infant deaths and consanguinity. [1] [3]
On examination, look for signs across systems: poor growth and wasted buttocks, an over-inflated chest with crackles or wheeze, digital clubbing in established lung disease, nasal polyps, abdominal distension and, in some, hepatomegaly. Early in life the examination can be entirely normal in a screen-detected infant, so a normal examination never excludes the diagnosis. [5] [3]
The single most useful bedside test is the sweat test, and its result must be interpreted against the reference thresholds rather than as a simple positive or negative. Bedside assessment therefore means deciding who needs a sweat test and arranging it at an accredited laboratory, because technique and sample adequacy strongly affect reliability. [11] [1]
Investigations
The sweat chloride test is the cornerstone investigation. Sweat is stimulated by pilocarpine iontophoresis, collected, and its chloride concentration measured in an accredited laboratory to strict quality standards. A chloride at or above sixty millimoles per litre is diagnostic when repeated, thirty to fifty-nine is intermediate and needs genetics and repeat testing, and below thirty makes CF unlikely. An adequate sweat sample and correct technique are essential to a reliable result. [11] [1]
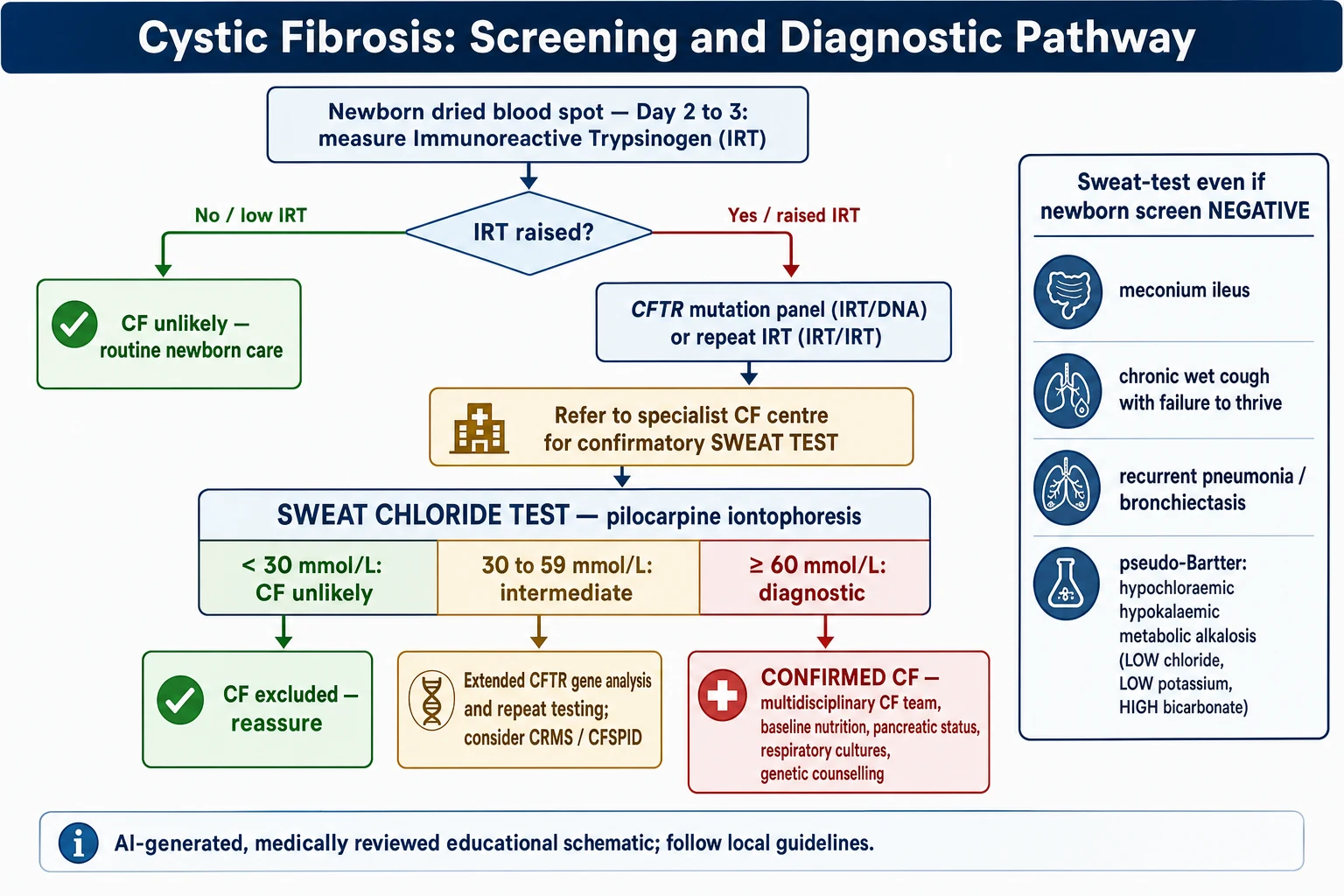
Newborn bloodspot screening is the population-level investigation. It begins by measuring immunoreactive trypsinogen, which is raised in most affected newborns, and then applies a second step — usually a CFTR mutation panel (IRT/DNA) or a repeat trypsinogen (IRT/IRT) — to select infants for a confirmatory sweat test. Screening finds most, but not all, affected infants, and its sensitivity depends on the mutation panel used. [8] [7]
CFTR genetic analysis confirms and refines the diagnosis. Finding two CF-causing mutations establishes CFTR dysfunction, guides prognosis, and increasingly determines eligibility for mutation-specific modulator therapy. Because standard panels test only common mutations, extended gene sequencing is used when the picture is suggestive but the panel is negative or incomplete. [6] [1]
The diagnostic pathway from suspicion to confirmation
Trigger: a positive newborn screen, or a clinical feature, or an affected sibling.
Refer to an accredited paediatric CF centre without delay.
Confirmatory sweat chloride test by pilocarpine iontophoresis, ensuring an adequate sample.
Interpret: below 30 unlikely, 30–59 intermediate, 60 or above diagnostic — repeat to confirm.
CFTR genetic analysis for confirmation, prognosis and modulator eligibility; extend gene testing if needed.
Confirm the diagnosis, or assign CRMS/CFSPID if results are equivocal, and start multidisciplinary care.
When the diagnosis is confirmed, baseline assessment at the CF centre defines the starting point: nutritional and pancreatic status with faecal elastase, respiratory cultures, and a review of growth and organ involvement. Nasal potential difference and intestinal current measurement are specialised tests of CFTR function reserved for genuinely equivocal cases in expert centres. [4] [1]
Management — Resuscitation
Cystic fibrosis is rarely an immediate resuscitation problem, but two presentations at diagnosis can be acutely dangerous and must be recognised and stabilised before the diagnostic workup proceeds. The first is meconium ileus in a newborn, and the second is severe salt-loss dehydration with pseudo-Bartter syndrome. Both need prompt correction alongside referral. [5] [1]
The newborn with meconium ileus presents with bilious vomiting, abdominal distension and failure to pass meconium, and needs resuscitation with fluids, nasogastric decompression and urgent surgical and neonatal review. Uncomplicated cases may respond to a contrast enema, while complicated ileus with perforation or atresia needs surgery. Meconium ileus is so strongly associated with CF that a sweat test and genetics should follow, whatever the screen shows. [5] [3]
Stabilising the acute CF presentation before confirmation
The child with pseudo-Bartter syndrome presents dehydrated with a hypochloraemic, hypokalaemic metabolic alkalosis and, characteristically, no history of vomiting to explain it. Resuscitate with isotonic saline to correct the sodium and chloride deficit, add potassium once urine flow is established, and recognise the biochemical pattern as a pointer to CF that warrants a sweat test once the child is stable. [1] [5]
Management — Definitive & Stepwise
The definitive step at diagnosis is not a drug but a pathway: confirm CFTR dysfunction, secure a genetic diagnosis, and hand the child promptly to a specialist multidisciplinary CF centre. Getting the child into structured CF care early is the intervention that changes outcome, because screened, early-treated children grow and breathe better than those diagnosed late. [10] [9]
Confirm the diagnosis first. Take every positive newborn screen and every clinically suspected child to an accredited CF centre for a confirmatory sweat chloride test, interpreted against the reference thresholds and repeated to confirm, with CFTR genetic analysis to establish the genotype. Do not begin lifelong CF care, with its burden and label, on a screen result alone. [2] [1]
CF SWEAT
Then start multidisciplinary care. Once confirmed, the child enters a CF team of paediatric respiratory and gastroenterology specialists, dietitians, physiotherapists, nurses and psychologists, with baseline nutritional, pancreatic and respiratory assessment and genetic counselling for the family. The detail of ongoing airway clearance, nutrition and modulator therapy belongs to the management topic; the diagnostic task is to reach this point quickly and correctly. [4] [1]
Genetic counselling is part of definitive management, not an afterthought. Confirm the parents' carrier status, explain the one-in-four recurrence risk, and offer counselling about reproductive options and cascade testing of relatives. Where a screen or diagnosis is equivocal, explain the CRMS or CFSPID label honestly and arrange structured follow-up rather than either firm reassurance or a firm CF diagnosis. [1] [12]
Specific Subtypes & Scenarios
CRMS and CFSPID is the scenario examiners love. An infant with a positive newborn screen but sweat chloride in the intermediate range and fewer than two clearly CF-causing mutations does not meet criteria for CF. This is labelled CFTR-related metabolic syndrome in North America, or CF screen-positive inconclusive diagnosis in Europe, and needs structured monitoring because a minority later evolve into CF. [2] [1]
Non-classic (atypical) cystic fibrosis presents later and milder, often pancreatic-sufficient, in a child or adult with bronchiectasis, recurrent pancreatitis, sinus disease or male infertility from congenital absence of the vas deferens. The sweat chloride may sit in the intermediate range, so extended CFTR genetics and specialist CFTR-function testing are often needed to reach or exclude a diagnosis. [3] [5]
Farrell 1997 — Wisconsin CF Neonatal Screening RCT — N Engl J Med (PMID 9395429)
Randomised controlled trial comparing screened (early-diagnosed) versus control (standard clinical diagnosis) newborns with CF
Key finding
Screened children had significantly better anthropometric nutritional status, showing a real benefit of early diagnosis on growth.
Practice change
Early diagnosis through newborn screening improves nutrition, underpinning universal bloodspot screening for CF.
The false-negative newborn screen is a critical trap. Screening misses some affected infants, especially those with rare mutations not on the local panel or with a normal first trypsinogen. A negative screen therefore never excludes CF in a child whose clinical picture fits, and the correct response to a suggestive presentation is a sweat test regardless of the screen. [7] [3]
The antenatal presentation completes the picture. Echogenic fetal bowel on ultrasound or a family history may prompt antenatal counselling and genetic testing, and a newborn with meconium ileus should be investigated for CF from birth. These scenarios test whether you can integrate the antenatal, screening and clinical routes into one coherent diagnostic plan. [1] [6]
Complications & Pitfalls
The complications relevant to diagnosis are the consequences of getting it wrong or getting it late. A missed or delayed diagnosis allows malnutrition, fat-soluble vitamin deficiency and progressive bronchiectasis to develop before treatment starts, and the Wisconsin trial showed that late diagnosis costs growth that early diagnosis preserves. Late diagnosis is the complication the whole screening system exists to prevent. [10] [9]
The dominant pitfalls cluster around the sweat test and the screen. The first is the technically inadequate sweat test: an insufficient sample or poor technique gives an unreliable result, so testing must be done in an accredited laboratory and repeated to confirm. The second is misreading an intermediate sweat chloride as either normal or diagnostic, when thirty to fifty-nine millimoles per litre demands genetics and repeat testing. [11] [1]
The third pitfall is over-trusting a negative newborn screen. Screening is not perfect, and a child with meconium ileus, failure to thrive with a wet cough, or pseudo-Bartter needs a sweat test even after a negative screen. The fourth is mishandling the equivocal result, either alarming a family with a firm CF diagnosis or falsely reassuring them, when the honest answer is a CRMS or CFSPID label with structured follow-up. [7] [2]
[4]Prognosis & Disposition
The prognostic message of this topic is that diagnosis timing itself shapes outcome. Children diagnosed early through screening and entered into specialist care have better nutrition, better preserved lung function and, over a lifetime, better survival than children diagnosed late once symptoms and organ damage have appeared. The randomised Wisconsin evidence anchors this claim for nutrition. [10] [9]
Disposition after a positive screen or a suggestive clinical picture is straightforward: refer promptly to an accredited paediatric CF centre for confirmation and, if confirmed, lifelong multidisciplinary care. A child with an intermediate or equivocal result is not discharged but enters structured CRMS or CFSPID surveillance, and a child with a genuinely negative sweat test and reassuring picture can be safely reassured while keeping the diagnosis in mind if new features appear. [2] [1]
Longer-term prognosis in confirmed CF has transformed with modern care and, for eligible genotypes, CFTR modulator therapy, but that story belongs to the management topic. From the diagnostic standpoint, the deliverable is early, accurate identification and prompt referral, because that is what buys the child the best possible starting point. [12] [4]
Special Populations
The infant with a positive newborn screen is the commonest special situation and needs prompt, calm handling: urgent referral, a confirmatory sweat test, and honest communication with anxious parents who often have a well-looking baby. Early salt supplementation and nutritional attention begin once the diagnosis is confirmed. [2] [1]
Children of non-European ancestry are a group in whom the diagnosis is easily delayed, because standard mutation panels are weighted toward European mutations and may not detect their CFTR variants. In these children a suggestive clinical picture should lead to a sweat test and extended gene sequencing rather than reliance on a negative panel-based screen. [6] [12]
Indigenous, rural, remote and disadvantaged children face the same disease with less access to accredited sweat-testing laboratories and specialist CF centres. Equity here means reliable referral pathways, telehealth links to CF centres, attention to salt loss in hot climates, and ensuring that a positive screen in a remote setting still reaches confirmatory testing without delay. [9] [7]
The child with an equivocal result (CRMS/CFSPID) is a special population in their own right, needing structured monitoring for evolving features, clear communication that avoids both false alarm and false reassurance, and a defined plan for when to re-test and when to reclassify as CF. [2] [1]
Evidence, Guidelines & Regional Differences
| Region | Guideline / source | Screening and diagnostic emphasis | Notes |
|---|---|---|---|
| ANZ | RCH Melbourne and state screening programs | Universal IRT-based newborn screen; confirmatory sweat test at a CF centre | Prompt referral; sweat test for clinical suspicion despite a negative screen |
| North America | CF Foundation 2017 consensus guidelines | Sweat chloride ≥ 60 diagnostic; CRMS for equivocal screens | Emphasises genetic confirmation and modulator eligibility |
| Europe / UK | ECFS best practice and newborn screening guidelines | IRT then DNA; CFSPID for inconclusive diagnosis | Panel must reflect local population; negative screen does not exclude CF |
| Low-resource | Clinical diagnosis where no screening exists | Failure to thrive, suppurative lung disease, salt loss | Access to accredited sweat testing is the limiting factor |
The evidence backbone you should be able to name starts with the Cystic Fibrosis Foundation 2017 consensus guidelines and their companion papers on diagnosis in screened and non-screened populations, which set the modern criteria of a clinical or screening trigger plus objective CFTR dysfunction. The ECFS best practice guidelines provide the European counterpart, and both harmonise the sweat chloride thresholds and the equivocal-result categories. [1] [2] [4]
For screening and its benefit, the Wisconsin randomised trial (Farrell) demonstrated the nutritional advantage of early diagnosis, and reviews of newborn screening confirm improvements in growth and, less consistently, respiratory outcomes. The CF Foundation newborn screening implementation guidance and the sweat-testing standards define how to run the programme and the test reliably. [10] [9] [8] [11]
The live areas of nuance are the choice of screening algorithm between IRT/DNA and IRT/IRT and next-generation-sequencing approaches, how best to manage and communicate the CRMS and CFSPID categories, and how to ensure equitable detection across ancestries when mutation panels are Eurocentric — good examples of an established framework still being refined. [12] [2]
Exam Pearls
And when you teach cystic fibrosis diagnosis, teach it as two converging paths — the newborn screen that catches most infants before symptoms, and the clinical alertness that catches the rest — both meeting at the sweat test and the CFTR genotype, so that no child with salty skin, a wet chest and poor growth slips through. [1] [9]
References
- [1]Farrell PM, White TB, Ren CL, et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr, 2017.PMID 28129811
- [2]Farrell PM, White TB, Howenstine MS, et al. Diagnosis of Cystic Fibrosis in Screened Populations. J Pediatr, 2017.PMID 28129810
- [3]Sosnay PR, White TB, Farrell PM, et al. Diagnosis of Cystic Fibrosis in Nonscreened Populations. J Pediatr, 2017.PMID 28129813
- [4]Castellani C, Duff AJA, Bell SC, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros, 2018.PMID 29506920
- [5]Elborn JS. Cystic fibrosis. Lancet, 2016.PMID 27140670
- [6]Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet, 2015.PMID 25404111
- [7]Grosse SD, Boyle CA, Botkin JR, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep, 2004.PMID 15483524
- [8]Comeau AM, Accurso FJ, White TB, et al. Guidelines for implementation of cystic fibrosis newborn screening programs: Cystic Fibrosis Foundation workshop report. Pediatrics, 2007.PMID 17272609
- [9]Dijk FN, Fitzgerald DA. The impact of newborn screening and earlier intervention on the clinical course of cystic fibrosis. Paediatr Respir Rev, 2012.PMID 23069119
- [10]Farrell PM, Kosorok MR, Laxova A, et al. Nutritional benefits of neonatal screening for cystic fibrosis. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. N Engl J Med, 1997.PMID 9395429
- [11]LeGrys VA, Yankaskas JR, Quittell LM, et al. Diagnostic sweat testing: the Cystic Fibrosis Foundation guidelines. J Pediatr, 2007.PMID 17586196
- [12]De Boeck K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr, 2020.PMID 31899933