Paeds · respiratory-sleep-and-airway
Cystic fibrosis: pulmonary and multidisciplinary management
Also known as CF lung disease management · Cystic fibrosis airway clearance and antibiotics · CFTR modulator therapy in children · Multidisciplinary cystic fibrosis care · Paediatric CF pulmonary care
Fellowship guide to the pulmonary and multidisciplinary management of cystic fibrosis in children: the CFTR channel defect that dehydrates the airway surface and starts a self-perpetuating cycle of mucus plugging, chronic infection and neutrophilic inflammation ending in bronchiectasis; the four pillars of daily airway clearance physiotherapy, mucoactive nebulisers such as dornase alfa and hypertonic saline, an anti-infective ladder from Pseudomonas eradication through cycled inhaled antibiotics and chronic azithromycin to intravenous therapy for exacerbations, and CFTR modulator therapy with elexacaftor-tezacaftor-ivacaftor; the nutrition, pancreatic enzyme and psychosocial work of the multidisciplinary team; infection segregation; recognising exacerbations, haemoptysis, pneumothorax, allergic bronchopulmonary aspergillosis and CF-related diabetes; and the ANZ, UK and North American approach to monitoring, transition and lung transplantation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the ten-year-old seen in the CF clinic: she is thin despite a big appetite, she has a loose productive cough most mornings, her last airway culture grew Pseudomonas, and her spirometry has slipped a little since last visit. Around her sits a team — a physiotherapist checking her airway clearance technique, a dietitian adjusting her enzymes and calories, a nurse reviewing her nebulisers, and a doctor deciding whether she is having an exacerbation. That scene is the whole of cystic fibrosis management: a chronic, multisystem disease held in check by daily preventive work rather than cured by any single treatment. [8] [9]
Cystic fibrosis is an autosomal recessive disease caused by mutations in the CFTR gene, whose protein is a chloride and bicarbonate channel at the surface of epithelial cells. When that channel fails, the fluid lining the airway becomes dehydrated, mucus becomes thick and sticky, and the lung loses its ability to clear inhaled organisms — so the pulmonary problem is fundamentally one of impaired mucociliary clearance leading to chronic infection. [9] [1]
Why this matters at fellowship level is that cystic fibrosis has been transformed within a generation. Median survival has risen from early childhood decades ago to the fifth decade and beyond, driven first by aggressive airway clearance, nutrition and infection control, and now by CFTR modulator drugs that correct the basic defect. The examiner wants a candidate who understands the disease mechanism, can structure the preventive management, and can recognise the complications and the emergencies. [8] [1]
Classification
The useful way to frame a child with cystic fibrosis is along three axes at once: which CFTR mutation they carry, because that predicts modulator eligibility; how advanced their lung disease is, because that drives the intensity of therapy; and what is growing in their airway, because chronic Pseudomonas changes the whole trajectory. [9] [1]
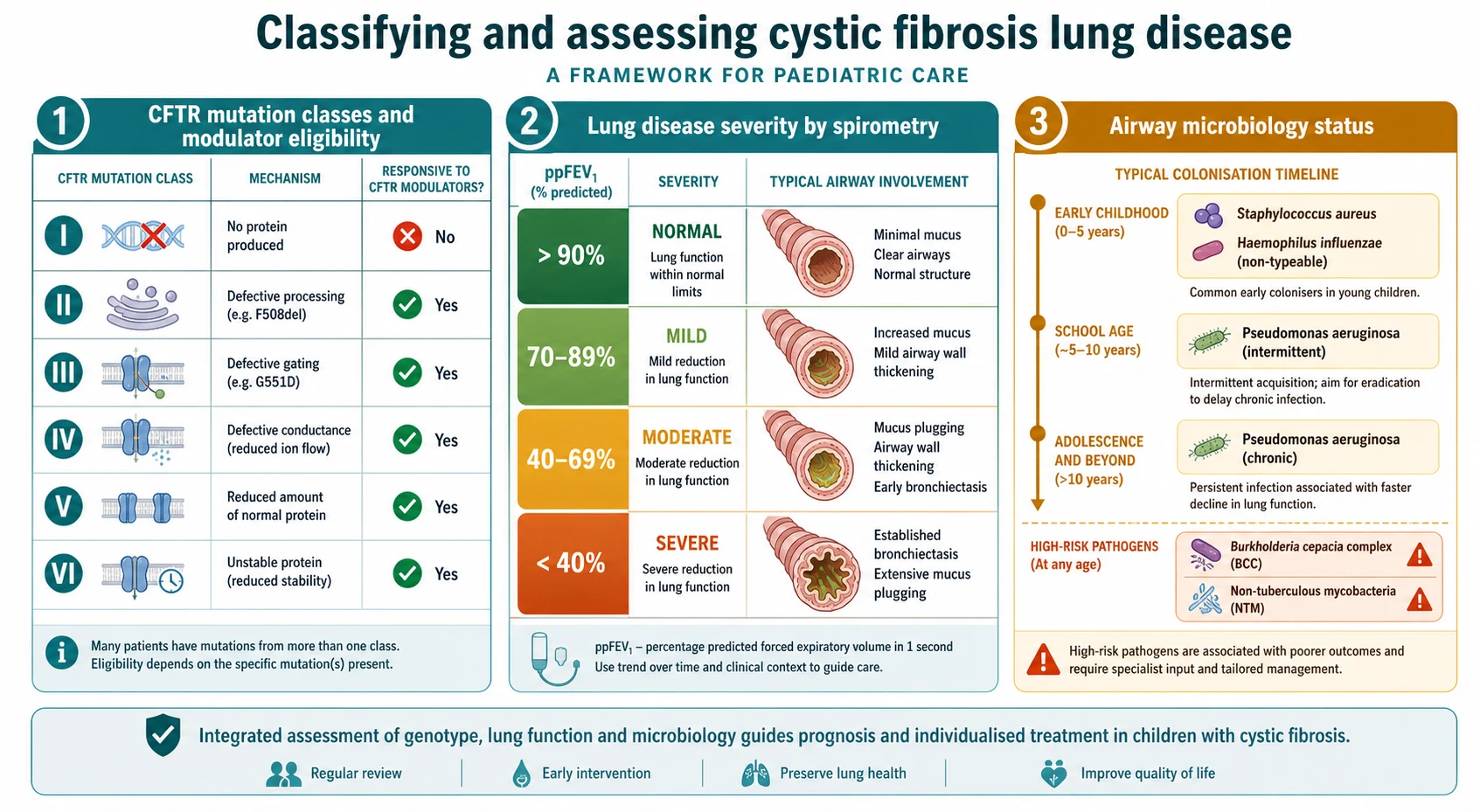
The first axis is the CFTR mutation class, which groups mutations by how they disable the protein: no protein made, defective processing of the protein so it never reaches the surface as in the common Phe508del, faulty gating as in G551D, reduced conductance, reduced quantity, and reduced stability. This classification matters because it predicts the type of CFTR modulator a child can respond to, from potentiators for gating defects to the corrector-potentiator combinations for processing defects. [2] [1]
The second axis is lung disease severity, judged mainly by spirometry once a child is old enough to perform it, and the third is airway microbiology. A child moves over years from early Staphylococcus aureus and Haemophilus influenzae, through first and intermittent Pseudomonas aeruginosa, to chronic Pseudomonas infection, and the isolation of Burkholderia cepacia complex or non-tuberculous mycobacteria marks a higher-risk course. [11] [7]
The numbers that anchor your viva
Epidemiology & Risk Factors
Cystic fibrosis is the commonest life-limiting autosomal recessive disease in populations of European ancestry, affecting roughly one in two and a half to three thousand live births, with a carrier frequency of about one in twenty-five. It is less common but not absent in other ethnic groups, and it occurs across all the populations seen in Australian, New Zealand, UK and North American practice. [9] [8]
The disease is inherited, so the main risk factor is genetic: two carrier parents have a one-in-four chance of an affected child with each pregnancy. The severity of lung disease, however, is shaped by more than the CFTR genotype alone, because modifier genes, the airway microbiome, adherence to therapy, nutrition and access to specialist care all influence how fast the lung deteriorates. [9] [8]
Within a diagnosed population, the strongest predictors of a worse pulmonary course are early and chronic Pseudomonas aeruginosa infection, poor nutritional status, frequent pulmonary exacerbations, and infection with Burkholderia cepacia complex or non-tuberculous mycobacteria. These are the levers the team pulls on, because eradicating early Pseudomonas, protecting nutrition and treating exacerbations promptly each measurably change the trajectory. [11] [7]
Pathophysiology
The teaching model runs from a broken channel to a destroyed airway, and it turns on one idea: without functioning CFTR, the airway surface dries out, mucus cannot be cleared, and the lung becomes a chronically infected, inflamed organ that slowly scars. Every treatment maps onto a step in this chain. [9] [1]
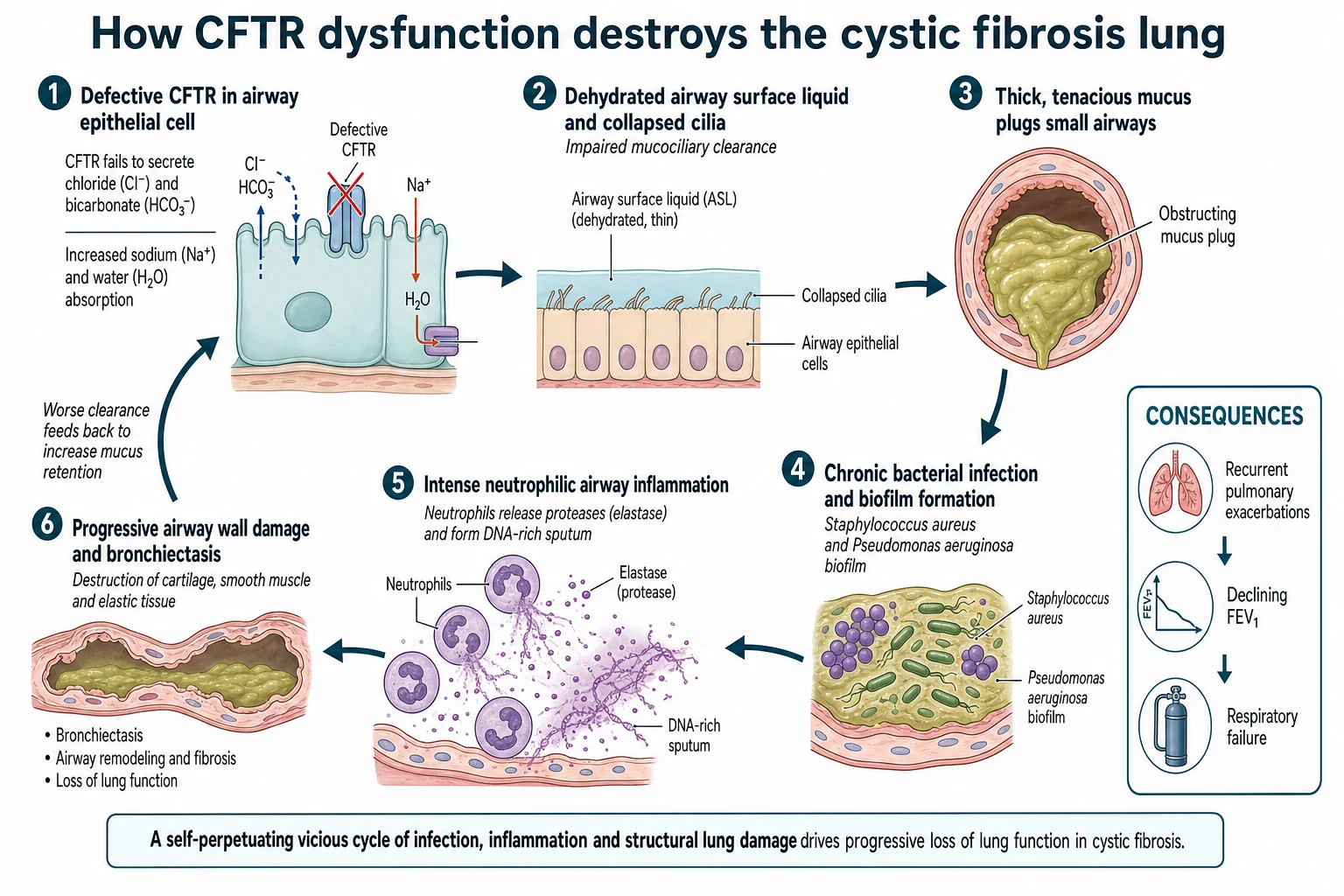
The primary defect is a failure of CFTR to secrete chloride and bicarbonate at the epithelial surface, with excess sodium and water absorption. The airway surface liquid becomes depleted and the mucus layer dehydrated, so the cilia can no longer sweep mucus up the airway. Impaired mucociliary clearance is the pivotal event, because it lets inhaled and aspirated organisms persist rather than being cleared. [9] [5]
Thick, retained mucus then becomes a niche for chronic bacterial infection, first with Staphylococcus aureus and Haemophilus and later, characteristically, with Pseudomonas aeruginosa, which forms protective biofilms. The host responds with an intense, largely neutrophilic inflammation, and the neutrophils release elastase and spill DNA that make the sputum even thicker and more damaging. [7] [4]
The cycle is self-perpetuating: infection and inflammation impair clearance further, and repeated injury destroys the airway wall to produce bronchiectasis, air trapping and, eventually, respiratory failure. Understanding this loop explains why management is preventive and daily rather than reactive, and why correcting the underlying CFTR defect with a modulator can change the whole downstream cascade. [1] [8]
Clinical Presentation
Most children in ANZ, UK and North American practice are diagnosed presymptomatically through newborn screening, so the pulmonary presentation the fellowship candidate manages is usually the established disease rather than the first clue. The typical picture is a chronic productive or loose cough, recurrent chest infections, and on examination coarse crackles, hyperinflation and, in advanced disease, finger clubbing. [9] [8]
The presentation that must be recognised quickly is the pulmonary exacerbation, because it is the event that drives lung decline. A child with an exacerbation has increased cough, more or more purulent sputum, breathlessness, tiredness, reduced appetite and weight, and a measurable fall in spirometry from their personal best. Fever is often mild or absent, so the diagnosis rests on the change from the child's usual state rather than on a single sign. [8] [7]
| Feature | Stable baseline | Exacerbation |
|---|---|---|
| Cough and sputum | Usual pattern for the child | Increased volume, purulence or frequency |
| Breathing and energy | At personal best | New breathlessness, fatigue, exercise intolerance |
| Weight and appetite | Stable on growth chart | Reduced appetite and weight loss |
| Spirometry | At or near personal best | Fall in FEV1 from best |
| Action | Continue maintenance therapy | Intensify airway clearance and start antibiotics |
Beyond the lungs, the disease declares itself in other systems that the multidisciplinary team manages in parallel: pancreatic insufficiency with steatorrhoea and poor growth, CF-related diabetes emerging in later childhood and adolescence, liver disease, sinus disease with nasal polyps, and male infertility. The pulmonary and nutritional problems are linked, because nutritional status and lung function track together over time. [9] [8]
Differential Diagnosis
Sort the differential into two questions: in the undiagnosed child with suppurative lung disease, is this cystic fibrosis or one of its mimics; and in the diagnosed child who deteriorates, is this a routine bacterial exacerbation or a specific complication that needs different treatment? The second question is the one the fellowship candidate faces most often. [9] [8]
Cystic fibrosis
the diagnosis
- Chronic productive cough, bronchiectasis
- Pancreatic insufficiency, poor growth
- Positive sweat test and CFTR mutations
- Pseudomonas colonisation over time
Primary ciliary dyskinesia
- Neonatal respiratory distress at term
- Chronic wet cough and bronchiectasis
- Situs inversus in about half
- Normal sweat test; abnormal cilia
Immunodeficiency
- Recurrent sinopulmonary infection
- Unusual or severe organisms
- Failure to thrive
- Low immunoglobulins or poor responses
CF exacerbation vs ABPA
- Exacerbation: bacterial, responds to antibiotics
- ABPA: wheeze, rising IgE, Aspergillus
- ABPA needs steroids and antifungal
- Missing ABPA worsens lung damage
In the undiagnosed child, the mimics of cystic fibrosis are the other causes of chronic suppurative lung disease and bronchiectasis: primary ciliary dyskinesia, immunodeficiency, recurrent aspiration and, in some settings, post-infectious bronchiectasis. The sweat test and CFTR genetic analysis separate cystic fibrosis from these, and pancreatic insufficiency with a positive sweat test is highly suggestive. [9] [8]
In the diagnosed child who deteriorates, the key distinctions are between an ordinary bacterial exacerbation and the specific complications that masquerade as one. Allergic bronchopulmonary aspergillosis presents with wheeze, a rising IgE and new infiltrates; a new atypical organism such as non-tuberculous mycobacteria can cause a decline that does not respond to standard antibiotics; and CF-related diabetes can drive weight loss and recurrent infections. Each needs a different treatment from a simple course of antibiotics. [8] [6]
Clinical & Bedside Assessment
Assessment in the CF clinic is structured and repeated at every visit, because the whole model depends on catching decline early. Take a focused history of cough, sputum, breathlessness, exercise tolerance, appetite and weight, adherence to airway clearance and nebulisers, and any recent symptoms of an exacerbation, then examine for work of breathing, chest signs, clubbing, nutritional state and growth. [8] [9]
Objective monitoring anchors the assessment. Spirometry at each visit, tracked against the child's personal best, is the single most useful measure of pulmonary status once a child can perform it, and a fall from best is the operational definition of decline. Airway cultures — cough swab, sputum or, in the young child, a bronchoalveolar lavage where indicated — guide antibiotic choice and detect new Pseudomonas. Weight, height and body mass index are recorded and plotted every visit. [8] [11]
Reading the clinic review
Emergency
Massive haemoptysis, pneumothorax or severe respiratory failure
The bedside review deliberately covers more than the chest, because cystic fibrosis is multisystem. The team screens for CF-related diabetes with an annual oral glucose tolerance test from about ten years of age, checks liver and bone health, reviews fat-soluble vitamin levels, and, just as importantly, assesses adherence and psychosocial wellbeing, because the daily treatment burden is heavy and adherence directly affects outcome. [9] [8]
Investigations
The core investigations are those that monitor and direct daily care rather than establish the diagnosis, which is usually already made. Spirometry tracks lung function trends, airway microbiology cultures direct antibiotics and flag new Pseudomonas, and inflammatory or infective markers help judge the severity of an exacerbation. These are repeated regularly rather than ordered once. [8] [11]
Imaging supports but does not replace clinical and functional monitoring. A chest radiograph shows hyperinflation, bronchial wall thickening and, later, bronchiectasis, and is used at diagnosis, during significant exacerbations and periodically for surveillance. Computed tomography is more sensitive for early structural change but carries a radiation cost, so it is used selectively. Lung clearance index from multiple-breath washout is a sensitive early marker of small-airway disease in young children. [8] [10]
Investigation of the deteriorating child is targeted at the differential. Rising total IgE, specific Aspergillus IgE and new infiltrates support allergic bronchopulmonary aspergillosis; repeated mycobacterial cultures detect non-tuberculous mycobacteria; an oral glucose tolerance test detects CF-related diabetes; and, where haemoptysis or pneumothorax is suspected, urgent imaging and specialist input take priority. Each test is chosen because it will change management. [8] [6]
Management — Resuscitation
Most cystic fibrosis care is planned and preventive, but a small number of presentations are genuine emergencies that a fellowship candidate must manage on the acute take. The two classic ones are massive haemoptysis and pneumothorax, and both are handled with a structured airway, breathing and circulation approach alongside early specialist involvement. [8] [9]
Massive haemoptysis in cystic fibrosis usually comes from a hypertrophied, high-pressure bronchial artery. Protect the airway, position the child with the bleeding side down if it is known, give oxygen, correct any coagulopathy, resuscitate the circulation, and arrange urgent bronchial artery embolisation, which is the definitive treatment for significant or recurrent bleeding. Withhold airway clearance physiotherapy and non-essential nebulisers during active major bleeding. [8] [9]
A pneumothorax presents with sudden pleuritic chest pain and breathlessness and can be life-threatening in a child with limited pulmonary reserve. Give oxygen, and decompress and drain a significant or tension pneumothorax as an emergency before arranging specialist respiratory and, sometimes, surgical management, because pneumothorax in cystic fibrosis has a high recurrence rate. A severe pulmonary exacerbation with respiratory failure needs oxygen, prompt intravenous antibiotics, airway clearance support and early consideration of non-invasive ventilation and intensive care. [8] [7]
Intravenous antibiotics for a severe Pseudomonas exacerbation
The principle in the acute setting is to treat aggressively and early rather than to wait, because a child with cystic fibrosis has less reserve than a previously well child and each severe exacerbation can cause a step-down in long-term lung function. Escalate to senior respiratory and intensive care support early for haemoptysis, pneumothorax or respiratory failure. [8] [7]
Management — Definitive & Stepwise
The definitive management of cystic fibrosis lung disease rests on four pillars delivered together and every day: airway clearance, mucoactive therapy, an anti-infective strategy, and CFTR modulator therapy, all coordinated by the multidisciplinary team alongside nutrition. Doing all four consistently, rather than reaching for any single treatment, is what preserves lung function over years. [8] [1]
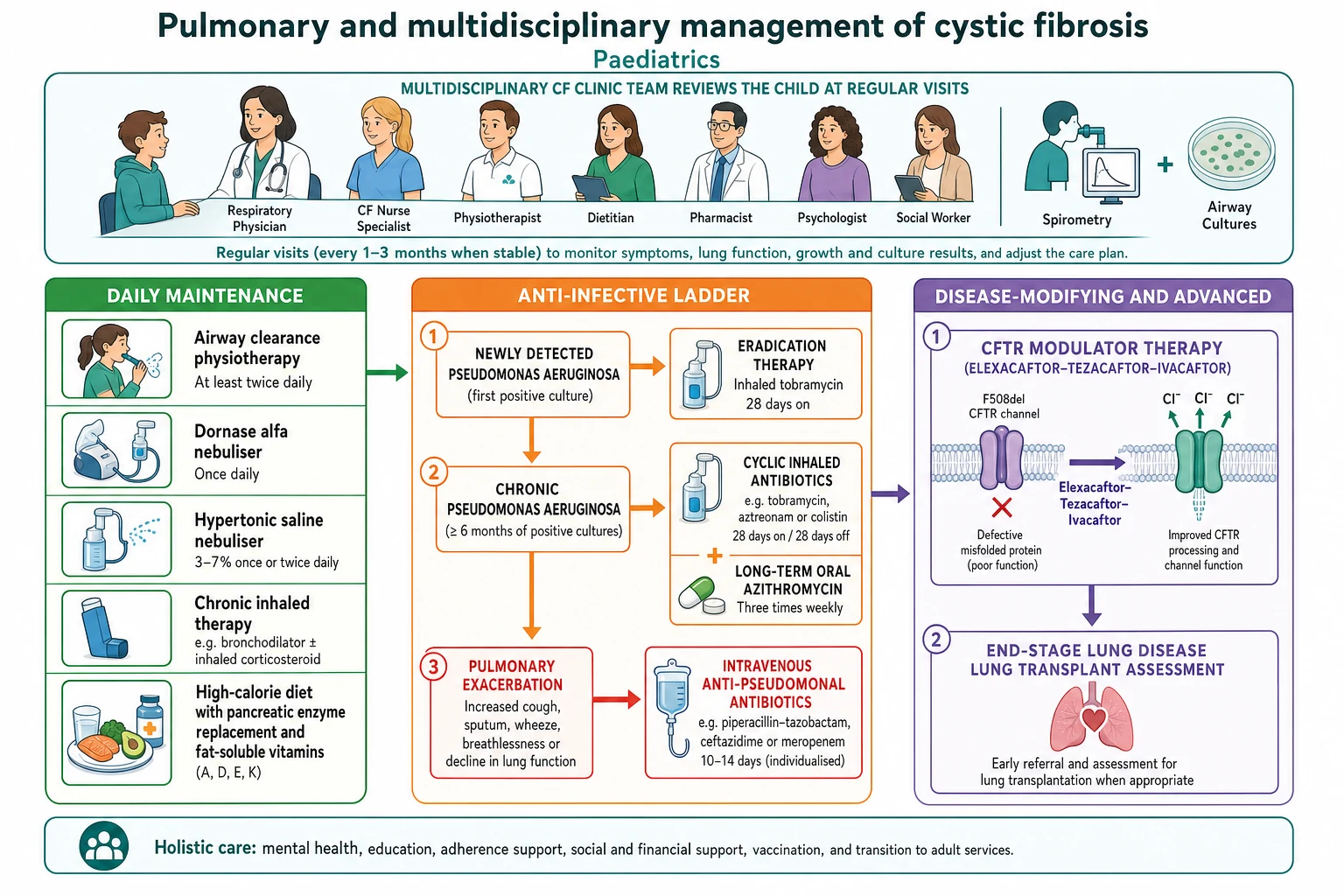
Airway clearance and mucoactive therapy are the daily foundation. Physiotherapy techniques — from percussion and postural drainage in the infant to active cycle of breathing, positive expiratory pressure devices and exercise in the older child — clear the retained mucus. Dornase alfa thins the DNA-laden sputum, and nebulised hypertonic saline rehydrates the airway surface; both improve clearance and lung function, and both are given regularly rather than only when unwell. [4] [5]
Maintenance mucoactive and inhaled therapy
The anti-infective strategy has two priorities: never let a first Pseudomonas isolate become chronic, and treat exacerbations promptly. A new Pseudomonas isolate triggers an eradication course, typically inhaled tobramycin with or without oral ciprofloxacin, which clears the organism in most children and delays chronic infection. Once infection is chronic, cycled inhaled antibiotics and long-term azithromycin suppress it, and exacerbations are treated with oral antibiotics for milder episodes or a course of two intravenous anti-pseudomonal agents for more severe ones. [11] [7]
The stepwise pulmonary pathway
Establish daily airway clearance physiotherapy and exercise, tailored to age and ability.
Add mucoactive nebulisers: dornase alfa and hypertonic saline to thin and hydrate secretions.
Eradicate the first Pseudomonas isolate promptly with inhaled tobramycin, aiming to prevent chronic infection.
Suppress chronic Pseudomonas with cycled inhaled antibiotics and long-term azithromycin.
Treat exacerbations early with oral or intravenous anti-pseudomonal antibiotics and intensified clearance.
Start CFTR modulator therapy for eligible mutations, and protect nutrition throughout with enzymes and calories.
CFTR modulator therapy is the transformative pillar, because it treats the defect rather than its consequences. Ivacaftor, a potentiator, restores gating in mutations such as G551D; the corrector-potentiator combination elexacaftor-tezacaftor-ivacaftor is highly effective for the large group of children with at least one Phe508del allele, improving lung function, reducing exacerbations and improving weight and quality of life, and it is now used down into younger childhood. Modulators are added to, not a replacement for, airway clearance, nutrition and infection management. [1] [2]
CF CARES
Specific Subtypes & Scenarios
The first Pseudomonas isolate is the scenario examiners love, because the right instinct is to act, not observe. A previously Pseudomonas-free child who grows the organism from a routine culture should be started on an eradication regimen straight away, because early treatment clears most children and prevents the transition to chronic infection that accelerates lung decline. [11] [7]
Allergic bronchopulmonary aspergillosis is the scenario where antibiotics are the wrong answer. A child with new wheeze, a rising total IgE, positive Aspergillus-specific IgE and new radiographic infiltrates who is not responding to antibacterial treatment has an allergic reaction to Aspergillus, and the treatment is oral corticosteroids with an antifungal such as itraconazole rather than escalating antibiotics. [8] [6]
Middleton 2019 — N Engl J Med (PMID 31697873)
Phase 3 randomised, double-blind, placebo-controlled trial in patients aged 12 and over with one Phe508del and one minimal-function allele
Key finding
The triple combination produced a large, sustained improvement in ppFEV1 and reduced pulmonary exacerbations compared with placebo.
Practice change
Elexacaftor-tezacaftor-ivacaftor extended highly effective modulator therapy to the majority of patients carrying at least one Phe508del allele.
CF-related diabetes is a distinct, common complication in later childhood and adolescence that worsens both lung function and nutrition. It is screened for with an annual oral glucose tolerance test and treated with insulin rather than oral agents, and good glycaemic control improves pulmonary outcomes. Non-tuberculous mycobacterial infection should be suspected when a child declines despite standard treatment, needs prolonged multidrug therapy, and can affect transplant candidacy. [9] [8]
The child too young for spirometry is managed by clinical assessment, weight and growth, airway cultures and, increasingly, the lung clearance index, and modulator eligibility now reaches into early childhood for some genotypes. The end-stage patient with severe, progressive disease despite full therapy is referred for lung transplant assessment, which can offer years of good-quality life. [10] [8]
Complications & Pitfalls
The pulmonary complications track the underlying process: recurrent exacerbations that each risk a step-down in lung function, chronic Pseudomonas and other resistant infections, bronchiectasis, haemoptysis from hypertrophied bronchial arteries, pneumothorax, allergic bronchopulmonary aspergillosis and, ultimately, respiratory failure. Each is anticipated and monitored for rather than merely treated once established. [8] [9]
Beyond the lungs, the disease is multisystem, and the team watches for pancreatic insufficiency and poor growth, CF-related diabetes, liver disease, distal intestinal obstruction syndrome, sinus disease and nasal polyps, low bone density, and, in adolescents, the psychosocial burden of a demanding chronic illness. Managing these well is inseparable from managing the lungs, because nutrition and lung function move together. [9] [8]
The dominant pitfalls are ones of delay and of missing a specific diagnosis. The first is observing rather than eradicating a first Pseudomonas isolate, which allows chronic infection to become established. The second is treating an allergic bronchopulmonary aspergillosis flare or a non-tuberculous mycobacterial decline as an ordinary bacterial exacerbation, so the child receives repeated antibiotics without improvement. [11] [8]
[8]The remaining pitfalls are under-treating nutrition, since letting weight fall drags lung function down with it, neglecting infection segregation, which allows cross-infection with Pseudomonas and especially Burkholderia between patients, and under-recognising the treatment burden and non-adherence, because a beautifully written plan that the family cannot sustain does not protect the lung. [9] [8]
Prognosis & Disposition
The prognosis of cystic fibrosis has been transformed and continues to improve. Median predicted survival, once measured in childhood, now reaches the fifth decade and beyond in well-resourced settings, driven by multidisciplinary care, aggressive infection and nutritional management, and, most recently, by highly effective CFTR modulator therapy that improves lung function and reduces exacerbations. The trajectory of an individual child depends heavily on preserving lung function early. [1] [8]
Disposition in cystic fibrosis is about the model of care rather than admit-versus-home for a single episode. Children are cared for in a specialist multidisciplinary CF centre with regular scheduled reviews, shared care with local services for families far from the centre, admission for intravenous antibiotics during significant exacerbations, and structured transition to adult CF services during adolescence. Emergencies such as haemoptysis and pneumothorax require immediate admission and specialist care. [8] [9]
The family's role is central, because the daily treatment burden of physiotherapy, nebulisers, enzymes and modulators falls on them, so education, psychosocial support and attention to adherence are part of the treatment. Clear plans for recognising and acting on an exacerbation, and for maintaining nutrition and clearance, keep the child out of hospital and preserve lung function. [8] [9]
Special Populations
The infant and very young child diagnosed by newborn screening is managed to protect the lung before symptoms appear, with airway clearance, close surveillance of growth and airway cultures, and prompt eradication of first infections, and for some genotypes modulator therapy now begins in early childhood. Nutrition is a priority from the start, because early nutritional status predicts later lung function. [9] [10]
The adolescent transitioning to adult care faces the double challenge of a heavy daily treatment burden and the developmental drive for independence, which puts adherence at risk. Structured transition programmes, attention to mental health, and the emergence of CF-related diabetes and reduced bone density in this age group all require the team's focus during these years. [9] [8]
Indigenous, rural, remote and socioeconomically disadvantaged children may live far from a specialist CF centre and face barriers to the daily therapies, nebuliser equipment and frequent reviews the disease demands. Equitable care depends on shared-care models with local services, telehealth review, support with the costs and logistics of treatment, and culturally appropriate education and communication. [9] [8]
Children without access to CFTR modulators, whether through age, genotype or, in many countries, cost and availability, still depend on the older pillars of airway clearance, mucoactive therapy, infection management and nutrition, which remain effective and essential. The global inequity in modulator access is a growing concern in the care of the disease. [8] [1]
Evidence, Guidelines & Regional Differences
| Region | Guideline / source | Emphasis | Notes |
|---|---|---|---|
| ANZ | TSANZ and RCH Melbourne | Newborn-screened diagnosis, specialist CF centres, eradication and segregation | Shared care and telehealth for rural and remote families |
| UK | NICE NG78 | Specialist multidisciplinary care, Pseudomonas eradication, modulator access | Strong emphasis on infection prevention and segregation |
| North America | CF Foundation guidelines | Chronic maintenance medications and airway clearance evidence base | Early and broad modulator adoption |
| Global | Access-limited settings | Older pillars of clearance, antibiotics and nutrition | Modulator cost and availability the limiting factor |
The evidence backbone you should be able to name starts with the mucoactive trials — the Pulmozyme study (Fuchs) for dornase alfa and the hypertonic saline trial (Elkins) — and the anti-infective trials — intermittent inhaled tobramycin (Ramsey) for chronic Pseudomonas, azithromycin (Saiman) as an anti-inflammatory, and the EPIC trial (Treggiari) on eradicating early Pseudomonas. Together they built the daily maintenance regimen that preserved lung function before modulators arrived. [4] [5] [7]
The modulator revolution is defined by a series of trials: ivacaftor for gating mutations (Ramsey), lumacaftor-ivacaftor for Phe508del homozygotes (Wainwright), and the triple combination elexacaftor-tezacaftor-ivacaftor (Middleton), extended to school-age children by Zemanick. The CF Foundation chronic-medication guidelines (Mogayzel) synthesise the maintenance evidence into practice. [1] [3] [10]
The live areas of nuance are how far modulator therapy will allow the older daily treatments to be simplified without losing lung function, the optimal management of non-tuberculous mycobacteria, and, above all, the global inequity in access to the modulators that now shape prognosis — a striking example of active care outpacing access. [1] [8]
Exam Pearls
And when you teach cystic fibrosis, teach it as the model of multidisciplinary chronic disease care — a broken channel that dehydrates the airway, a preventive daily regimen that keeps the cycle in check, and a modulator revolution that now corrects the defect itself — while never losing your alertness to the first Pseudomonas isolate, the exacerbation, and the haemoptysis or pneumothorax that turns a chronic illness into an emergency. [8] [1]
References
- [1]Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med, 2019.PMID 31697873
- [2]Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med, 2011.PMID 22047557
- [3]Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med, 2015.PMID 25981758
- [4]Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med, 1994.PMID 7503821
- [5]Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med, 2006.PMID 16421364
- [6]Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA, 2003.PMID 14519709
- [7]Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med, 1999.PMID 9878641
- [8]Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med, 2013.PMID 23540878
- [9]Farrell PM, White TB, Ren CL, et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr, 2017.PMID 28129811
- [10]Zemanick ET, Taylor-Cousar JL, Davies J, et al. A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am J Respir Crit Care Med, 2021.PMID 33734030
- [11]Treggiari MM, Retsch-Bogart G, Mayer-Hamblett N, et al. Comparative efficacy and safety of 4 randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Arch Pediatr Adolesc Med, 2011.PMID 21893650