Behçet's Disease
Behçet's disease (BD) is a chronic, relapsing, multisystem inflammatory disorder characterised by recurrent oral and gen... MRCP exam preparation.
What matters first
Behçet's disease (BD) is a chronic, relapsing, multisystem inflammatory disorder characterised by recurrent oral and gen... MRCP exam preparation.
Posterior uveitis (sight-threatening)
5 Jan 2026
Generated educational material; verify before clinical use.
Visible references section
See the concept before reading it
Study the key anatomy, imaging, and decision pathways as full teaching plates.
Clinical board
A visual summary of the highest-yield teaching signals on this page.
Urgent signals
Safety-critical features pulled from the topic metadata.
- Posterior uveitis (sight-threatening)
- Neurological involvement (parenchymal or vascular)
- Large vessel disease (arterial aneurysm)
- GI perforation
Exam focus
Current exam surfaces linked to this topic.
- MRCP
Linked comparisons
Differentials and adjacent topics worth opening next.
- Herpes Simplex Virus Infection
- Recurrent Aphthous Stomatitis
Content status and exam context
This page is AI-generated educational content. It may contain errors or omissions and is not a substitute for current guidelines, local protocols, senior clinical judgement, or professional medical advice.
MedVellum does not claim an individual clinician reviewer, board certification, or professional credential for this page unless a future version names a real, verifiable reviewer.
Clinical explanation and evidence
Behçet's Disease
1. Clinical Overview
Summary
Behçet's disease (BD) is a chronic, relapsing, multisystem inflammatory disorder characterised by recurrent oral and genital ulceration, ocular inflammation, and cutaneous lesions. First described by Turkish dermatologist Hulusi Behçet in 1937, it represents a variable-vessel vasculitis that can affect arteries and veins of all sizes. The disease follows a relapsing-remitting course with potential involvement of multiple organ systems including the eyes, nervous system, vascular system, and gastrointestinal tract. [1,2]
The pathogenesis involves a complex interplay of genetic susceptibility (particularly HLA-B51), environmental triggers, and dysregulated innate and adaptive immunity with neutrophil hyperactivity and Th1/Th17 predominance. There is no single diagnostic test for BD; diagnosis relies on clinical criteria, most commonly the International Criteria for Behçet's Disease (ICBD) or the older International Study Group (ISG) criteria. [3,4]
Management is tailored to organ involvement and disease severity. Mucocutaneous manifestations typically respond to colchicine and topical corticosteroids. Sight-threatening ocular disease, neurological involvement, and major vascular complications require prompt systemic immunosuppression with corticosteroids, conventional immunosuppressants (azathioprine, cyclosporine), and increasingly anti-TNF biological agents. Early recognition and aggressive treatment of organ-threatening disease is essential to prevent irreversible damage and improve long-term outcomes. [5,6]
Key Facts
- Definition: Systemic variable-vessel vasculitis characterised by recurrent oral ulcers, genital ulcers, ocular inflammation, and skin lesions
- Incidence: Highly variable; highest along the ancient Silk Road (Turkey 80-370 per 100,000; Iran 16-100 per 100,000; Japan 7-14 per 100,000; rare in Northern Europe and North America less than 1 per 100,000) [7,8]
- Peak Demographics: Onset typically in the third to fourth decade (20-40 years); slight male predominance overall, but disease more severe in males particularly for ocular and neurological involvement [9]
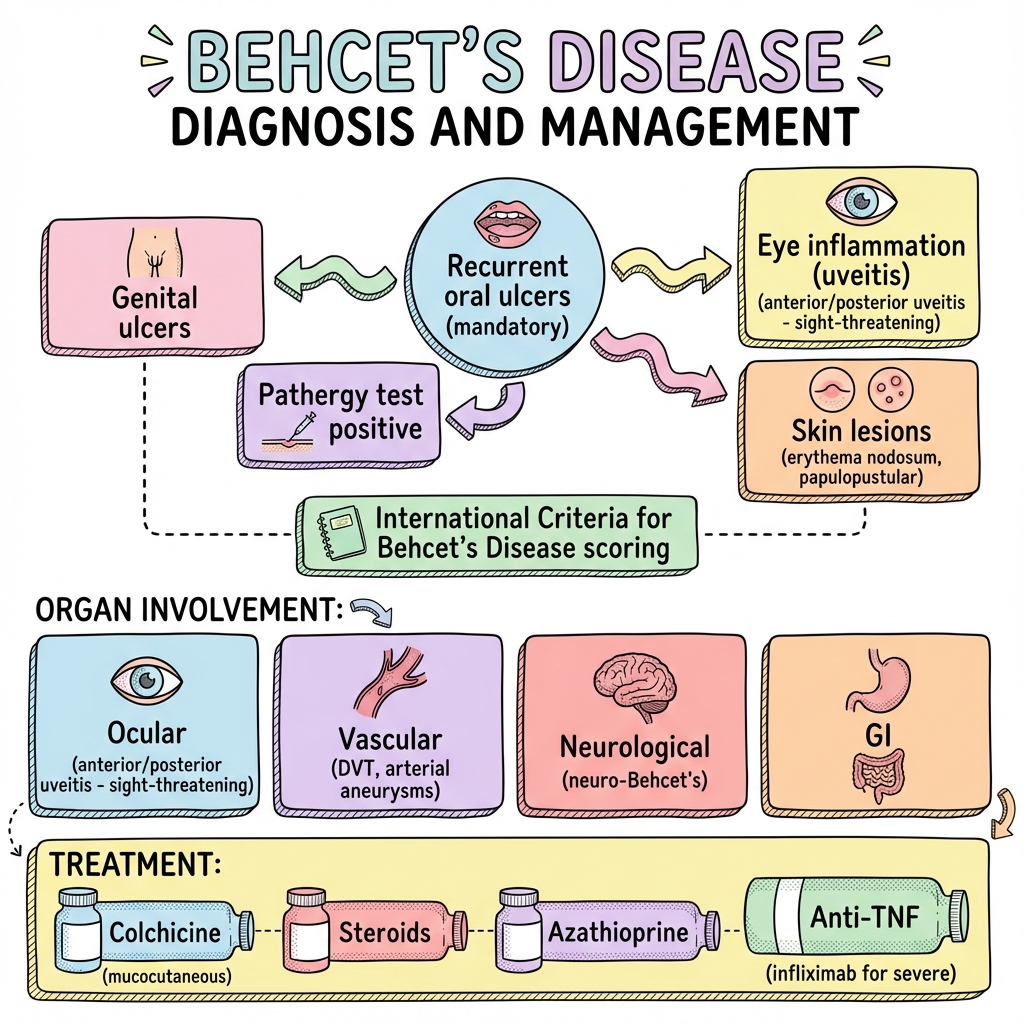
- Pathognomonic Features: Recurrent oral aphthous ulceration (mandatory criterion) plus genital ulcers, ocular lesions, skin lesions, or positive pathergy test
- Gold Standard Investigation: Clinical diagnostic criteria (ICBD or ISG); no definitive laboratory test
- First-line Treatment: Colchicine for mucocutaneous disease; systemic corticosteroids plus azathioprine or anti-TNF agents for organ-threatening manifestations
- Prognosis: Variable; eye disease is the major cause of morbidity; mortality increased with vascular and neurological involvement; 10-year survival > 90% in most cohorts [10]
Clinical Pearls
Diagnostic Pearl: To meet ISG criteria, oral ulcers must recur at least 3 times in a 12-month period. The ICBD criteria are more sensitive (94% vs 85%) but slightly less specific than ISG criteria. [11]
Emergency Pearl: Posterior segment uveitis (retinal vasculitis, vitritis) is sight-threatening and requires urgent ophthalmology assessment and systemic immunosuppression within 24 hours to prevent irreversible visual loss. [12]
Treatment Pearl: Anti-TNF agents (particularly infliximab and adalimumab) have revolutionised treatment of refractory ocular, neurological, and vascular Behçet's disease, with superior efficacy compared to conventional immunosuppressants in multiple RCTs. [13,14]
Geographic Pearl: The pathergy test is positive in 50-70% of Turkish patients but only 10-20% in Western European populations, limiting its diagnostic utility outside endemic regions. [15]
2. Epidemiology
Behçet's disease shows striking geographic variation in prevalence, following the historic Silk Road trade route from Eastern Asia through the Middle East to the Mediterranean basin. This distribution pattern has led to the alternative name "Silk Road Disease."
Geographic Distribution
| Region | Prevalence (per 100,000) | Notes | Reference |
|---|---|---|---|
| Turkey | 80-370 | Highest worldwide prevalence | [7] |
| Iran | 16-100 | High endemic area | [16] |
| Japan | 7-14 | Well-characterised cohorts | [17] |
| Korea | 3-4 | Lower than other Asian countries | [18] |
| Saudi Arabia | 20 | Variable across Middle East | [8] |
| Mediterranean (Greece, Italy) | 2-15 | Southern European distribution | [8] |
| Northern Europe (UK, Germany) | less than 1 | Very rare | [7] |
| North America (USA) | less than 1 | Mostly in immigrants | [8] |
Demographics
Age of Onset: The disease typically manifests in the third or fourth decade of life, with mean age at diagnosis between 25-35 years. Juvenile-onset BD (before age 16) accounts for approximately 3-26% of cases depending on the population studied, and tends to have a more severe course. [19,20]
Sex Distribution: Overall male-to-female ratio is approximately 1.2-2:1 in Middle Eastern and Asian populations. However, disease severity differs significantly by sex—males tend to have more severe ocular involvement, posterior uveitis, vascular complications, and neurological disease. Females may have more frequent genital ulceration and arthritis. [9]
Ethnicity: HLA-B51 prevalence parallels disease distribution. In Turkish populations, HLA-B51 is present in 50-70% of BD patients compared to 10-30% of controls. In Japanese BD patients, prevalence is 50-60%. In Northern European BD patients, HLA-B51 prevalence is lower (10-30%). [21]
Temporal Trends
Recent studies suggest stable or slightly decreasing incidence in endemic regions (Japan, Turkey) over the past two decades, possibly related to improved hygiene and reduced exposure to infectious triggers. Conversely, increased recognition and diagnosis may be occurring in Western countries. [22,23]
3. Aetiology & Pathophysiology
The aetiology of Behçet's disease remains incompletely understood but involves a complex interaction between genetic predisposition, environmental triggers, and immune dysregulation affecting both innate and adaptive immunity.
Genetic Factors
HLA-B51 Association: The strongest genetic association is with HLA-B51, particularly the HLA-B*51:01 allele. Meta-analyses demonstrate an odds ratio of 5.78-6.7 for disease development in HLA-B51 carriers. However, HLA-B51 accounts for only approximately 20% of genetic susceptibility, indicating substantial heterogeneity. [24,25]
The mechanism by which HLA-B51 predisposes to BD is uncertain. Proposed mechanisms include:
- Molecular mimicry with microbial antigens
- Presentation of disease-specific autoantigenic peptides
- Altered recognition of self vs non-self antigens
- Interaction with natural killer cell receptors affecting immune regulation [26]
Non-HLA Associations: Genome-wide association studies (GWAS) have identified multiple non-HLA susceptibility loci including:
- IL10, IL23R/IL12RB2 (Th1/Th17 polarisation)
- IL1 gene cluster (autoinflammatory pathways)
- ERAP1 (antigen processing)
- CCR1, STAT4, KLRC4 (immune regulation)
- MEFV (familial Mediterranean fever gene; modifier of disease severity) [27,28]
Environmental Triggers
Several environmental factors have been implicated:
Infectious Agents: Streptococcus sanguinis (oral flora) has been particularly implicated, with increased immune responses to streptococcal antigens and cross-reactivity with human heat shock proteins (HSP). Herpes simplex virus, Helicobacter pylori, and various other pathogens have also been studied but without conclusive evidence. [29]
Microbiome: Alterations in oral and gut microbiota composition have been reported in BD patients, with decreased diversity and altered Firmicutes/Bacteroidetes ratios suggesting a potential pathogenic role. [30]
Immunopathogenesis
Innate Immunity Dysregulation:
Neutrophil hyperactivity is a hallmark of BD pathogenesis:
- Enhanced chemotaxis and phagocytic activity
- Increased superoxide production and oxidative burst
- Elevated neutrophil extracellular trap (NET) formation
- Overexpression of neutrophil adhesion molecules (CD11b, CD18) [31]
Monocyte and dendritic cell dysfunction contributes through:
- Excessive IL-1β, TNF-α, and IL-6 production
- Enhanced Toll-like receptor (TLR) signalling
- Activation of inflammasome pathways (particularly NLRP3) [32]
Adaptive Immunity Dysregulation:
Th1 and Th17 polarisation predominates:
- Elevated IL-17A, IL-17F, IL-21, IL-22 levels in serum and tissues
- Increased frequency of circulating Th17 and Th1 cells
- IL-23/IL-17 axis amplification driving chronic inflammation
- Reduced regulatory T cell (Treg) numbers and function [33,34]
Gamma-delta (γδ) T cells are expanded in BD and produce high levels of pro-inflammatory cytokines including IFN-γ and IL-17. [35]
Endothelial Dysfunction:
Vasculitis in BD results from:
- Direct endothelial cell activation and damage
- Increased adhesion molecule expression (VCAM-1, ICAM-1, E-selectin)
- Enhanced leukocyte-endothelial interactions
- Procoagulant state with elevated von Willebrand factor and reduced protein C/S
- Oxidative stress and nitric oxide imbalance [36]
Cytokine Network:
Key cytokines implicated in BD pathogenesis:
- TNF-α: Central role; therapeutic target for anti-TNF biologics
- IL-1β: Drives autoinflammatory features; potential therapeutic target
- IL-17: Th17 signature cytokine; correlates with disease activity
- IL-6: Trans-signalling contributes to systemic inflammation
- IL-10: Reduced anti-inflammatory cytokine levels
- IFN-γ: Th1 polarisation marker [37]
4. Clinical Presentation
Behçet's disease is characterised by episodic inflammatory attacks involving multiple organ systems. The clinical course is typically relapsing-remitting with variable severity and frequency of exacerbations.
Mucocutaneous Manifestations
Oral Ulceration (97-100% of patients):
- Mandatory criterion for all diagnostic classifications
- Recurrent painful aphthous ulcers of the oral mucosa
- Typically 2-10mm diameter; can be larger (major aphthae)
- Affect buccal mucosa, tongue, lips, gingiva, palate, tonsils
- Usually heal within 1-2 weeks without treatment
- Often the initial manifestation, preceding other features by years
- Morphologically identical to common aphthous ulcers, creating diagnostic challenge [1,38]
Genital Ulceration (60-90%):
- Morphologically similar to oral ulcers but typically more painful
- Males: scrotum (most common), penis, inguinal region
- Females: vulva, vagina, cervix
- Typically deeper than oral ulcers and often heal with scarring
- May cause significant pain, dyspareunia, and psychosocial distress
- Strongly suggestive of BD when combined with recurrent oral ulcers [38]
Cutaneous Lesions (40-90%):
- Erythema nodosum-like lesions: Tender subcutaneous nodules, typically on lower extremities; most common cutaneous manifestation
- Papulopustular lesions: Acneiform lesions on trunk and extremities; sterile folliculitis
- Superficial thrombophlebitis: Tender subcutaneous cords, particularly lower limbs
- Sweet's syndrome-like lesions: Painful erythematous plaques (rare)
- Pyoderma gangrenosum-like ulcers: Deep ulcerations (rare) [39]
Pathergy Phenomenon (30-70% endemic regions; 10-20% Western populations):
- Hyperreactivity of skin to minor trauma (needle prick)
- Positive test: development of erythematous papule or pustule > 2mm at needle prick site after 24-48 hours
- Standardised testing: 20-gauge needle, intradermal insertion to 5mm depth, forearm site
- Specificity varies by population; less reliable in non-endemic regions
- Can occur spontaneously (needle sticks, shaving, vaccinations) [15,40]
Ocular Manifestations (30-70%)
Eye disease is the major cause of morbidity in BD and can lead to blindness if untreated. Ocular involvement is more common and severe in males and young patients.
Anterior Uveitis (50-80% of ocular BD):
- Acute anterior chamber inflammation with hypopyon (layered white cells in anterior chamber)
- Hypopyon is classic but occurs in less than 30% of anterior uveitis cases
- Presents with eye pain, photophobia, blurred vision, conjunctival injection
- Generally responds well to topical and systemic corticosteroids [12]
Posterior Uveitis and Panuveitis (50-80%):
- Retinal vasculitis: Inflammatory occlusive vasculitis affecting retinal vessels
- Vitritis: Inflammation of vitreous humor causing floaters and reduced vision
- Optic disc swelling and atrophy: Can cause permanent visual loss
- Retinal infiltrates, haemorrhages, exudates: Multiple patterns possible
- Macular oedema: Common cause of visual impairment
- Posterior segment involvement is the major threat to vision and requires aggressive systemic immunosuppression [12,41]
Complications of Ocular Disease:
- Cataract formation (from recurrent inflammation or corticosteroid use)
- Glaucoma (secondary to inflammation or corticosteroids)
- Retinal vein occlusion and ischaemia
- Optic atrophy
- Permanent vision loss in 10-25% of patients despite treatment [42]
Vascular Manifestations (7-40%)
BD can affect arteries and veins of all sizes, representing a true "variable vessel vasculitis."
Venous Thrombosis (15-40% of vascular BD):
- Deep vein thrombosis (DVT): lower extremity most common
- Superficial thrombophlebitis
- Superior/inferior vena cava thrombosis (Budd-Chiari syndrome when hepatic veins involved)
- Cerebral venous sinus thrombosis (see neurological manifestations)
- Characteristically adherent "sticky" thrombi with low risk of embolisation [43,44]
Arterial Disease (3-10%):
- Aneurysm formation: Pulmonary artery (most common and dangerous), aorta, peripheral arteries
- Arterial thrombosis: Less common than venous; can affect any arterial bed
- Pulmonary artery aneurysm: Life-threatening complication; presents with haemoptysis, chest pain; high mortality if ruptures
- Peripheral arterial aneurysms: Femoral, popliteal, subclavian arteries
- Arterial disease carries worse prognosis than venous thrombosis [43,45]
Neurological Manifestations (5-30%)
Neurological involvement ("neuro-Behçet's") is classified into parenchymal and non-parenchymal (vascular) subtypes.
Parenchymal Neuro-Behçet's (70-80% of neurological BD):
- Brainstem involvement: Most characteristic; ataxia, dysarthria, dysphagia, cranial nerve palsies, pyramidal signs
- Meningoencephalitis: Headache, confusion, behavioural changes, fever
- Hemispheric involvement: Hemiparesis, hemisensory loss, seizures
- Spinal cord involvement: Myelopathy, transverse myelitis (rare)
- Neuropsychiatric manifestations: Cognitive impairment, personality changes, psychosis
- MRI typically shows brainstem, basal ganglia, or hemispheric white matter lesions
- CSF shows lymphocytic pleocytosis, elevated protein, normal glucose [46,47]
Non-Parenchymal (Vascular) Neuro-Behçet's (20-30%):
- Cerebral venous sinus thrombosis: Superior sagittal sinus most common; presents with headache, papilloedema, seizures
- Intracranial aneurysms: Rare but life-threatening
- Generally better prognosis than parenchymal disease [46]
Gastrointestinal Manifestations (5-60%)
GI involvement varies dramatically by geography (more common in Japan, Korea) and can mimic inflammatory bowel disease.
Presentations:
- Ileocaecal ulceration (most common site; right lower quadrant pain)
- Oesophageal, gastric, duodenal, or colonic ulceration
- Deep, discrete, oval or round ulcers with sharp margins
- Complications: Perforation, fistula formation, haemorrhage, strictures
- Differentiation from Crohn's disease can be challenging; BD ulcers tend to be fewer in number, larger, and deeper [48,49]
Musculoskeletal Manifestations (40-70%)
- Arthralgia: Very common; non-erosive, non-deforming
- Arthritis: Typically oligoarticular, affecting knees, ankles, wrists, elbows
- Self-limiting, lasting days to weeks
- No radiographic erosions or joint destruction
- Sacroiliitis occasionally reported [50]
Other Manifestations
Cardiac (1-5%): Pericarditis, myocarditis, endocarditis (aortic/mitral valve), endomyocardial fibrosis, coronary arteritis, intracardiac thrombus [51]
Renal (rare): Glomerulonephritis (various types), renal artery stenosis, IgA nephropathy, amyloidosis (AA type in chronic disease) [52]
Pulmonary (5-10%): Pulmonary artery aneurysm, pulmonary artery thrombosis, pulmonary infarction, pleuritis, fibrosing alveolitis [45]
Epididymitis (5-10% of males): Unilateral or bilateral; can mimic infection or malignancy [53]
5. Differential Diagnosis
The diagnosis of Behçet's disease is clinical and often delayed due to the relapsing-remitting nature and overlap with other conditions. Key differentials to consider:
1. Recurrent Aphthous Stomatitis (RAS)
Key Distinguishing Features:
- Isolated recurrent oral ulcers without systemic features
- Very common (10-20% of general population)
- No genital ulcers, ocular inflammation, or systemic manifestations
- Pathergy test negative
- Diagnosis of BD requires systemic features beyond oral ulcers alone [54]
2. Herpes Simplex Virus (HSV) Infection
Key Distinguishing Features:
- Genital ulcers typically multiple, clustered vesicles that rupture
- Vesicular stage usually evident (not seen in BD)
- Tingling/burning prodrome common
- Viral culture, PCR, or direct fluorescent antibody positive for HSV
- Recurrent episodes can mimic BD genital ulcers
- HSV serology and viral detection essential to exclude [55]
3. Crohn's Disease
Key Distinguishing Features:
- Chronic diarrhoea, abdominal pain, perianal disease
- Oral aphthae can occur but less prominent than in BD
- Colonoscopy: skip lesions, cobblestoning, longitudinal ulcers
- Histology: transmural inflammation, non-caseating granulomas
- Positive inflammatory markers
- GI manifestations of BD can be indistinguishable; diagnosis challenging in Asian populations where both are common [48,49]
4. Systemic Lupus Erythematosus (SLE)
Key Distinguishing Features:
- Oral ulcers typically painless in SLE (painful in BD)
- Malar rash, photosensitivity, discoid lesions
- Positive ANA, anti-dsDNA, low complement
- Renal involvement common in SLE (rare in BD)
- Arthritis in SLE can be erosive/deforming (non-erosive in BD)
- SLE predominantly affects females; BD more equal or male predominance [56]
5. Sarcoidosis
Key Distinguishing Features:
- Multi-system granulomatous disease
- Bilateral hilar lymphadenopathy, pulmonary infiltrates
- Skin lesions: lupus pernio, erythema nodosum, papules
- Anterior uveitis in sarcoid typically granulomatous with "mutton-fat" keratic precipitates
- Elevated ACE, hypercalcaemia
- Non-caseating granulomas on biopsy
- Oral ulcers rare in sarcoidosis [57]
6. Other Vasculitides
Granulomatosis with Polyangiitis (GPA):
- Upper and lower respiratory tract involvement
- Necrotising glomerulonephritis
- c-ANCA/PR3 antibodies positive
- Oral/nasal ulceration but more destructive than BD [58]
Polyarteritis Nodosa (PAN):
- Medium-vessel arterial vasculitis
- Renal and GI involvement common
- No venous thrombosis (key difference from BD)
- Aneurysms but different distribution [58]
7. Reactive Arthritis (Reiter's Syndrome)
Key Distinguishing Features:
- Triad: arthritis, urethritis, conjunctivitis
- Follows GI or GU infection
- Painless oral ulcers, balanitis circinata
- HLA-B27 association
- Self-limited course in most cases [59]
8. MAGIC Syndrome (Mouth and Genital Ulcers with Inflamed Cartilage)
- Overlap syndrome between BD and relapsing polychondritis
- Auricular and nasal cartilage inflammation
- Rare [60]
9. Sweet's Syndrome (Acute Febrile Neutrophilic Dermatosis)
Key Distinguishing Features:
- Painful erythematous plaques and nodules
- Fever, neutrophilia
- Dermal neutrophilic infiltrate on histology
- Usually lacks genital ulcers and uveitis
- Can be associated with malignancy or medications [61]
| Differential | Oral Ulcers | Genital Ulcers | Uveitis | Pathergy | Key Distinguishing Feature |
|---|---|---|---|---|---|
| Behçet's Disease | +++ (recurrent, painful) | +++ (scarring) | ++ (anterior/posterior) | +/- (region-dependent) | Multi-system vasculitis |
| RAS | +++ | - | - | - | Isolated oral ulcers only |
| HSV | + | ++ (vesicular) | - | - | Vesicles, PCR/culture positive |
| Crohn's Disease | + | + (perianal) | + (anterior) | - | Transmural bowel inflammation, granulomas |
| SLE | + (painless) | - | - | - | ANA+, photosensitivity, renal disease |
| Sarcoidosis | - | - | + (granulomatous) | - | Hilar lymphadenopathy, granulomas |
6. Investigations
There is no specific diagnostic laboratory test for Behçet's disease. Diagnosis is clinical based on validated criteria. Investigations are used to:
- Support clinical diagnosis
- Assess organ involvement and complications
- Exclude mimicking conditions
Diagnostic Criteria
International Criteria for Behçet's Disease (ICBD) - 2014 [11]: More sensitive than ISG criteria (94% vs 85% sensitivity)
| Clinical Feature | Points |
|---|---|
| Oral aphthosis | 2 |
| Genital aphthosis | 2 |
| Ocular lesions | 2 |
| Skin lesions | 1 |
| Vascular manifestations | 1 |
| Pathergy test positive | 1 |
| Neurological manifestations | 1 |
Score ≥4 points = Behçet's Disease diagnosis
International Study Group (ISG) Criteria - 1990 [62]: More specific but less sensitive than ICBD
Mandatory criterion:
- Recurrent oral ulceration (at least 3 episodes in 12 months)
Plus 2 of the following:
- Recurrent genital ulceration
- Eye lesions (anterior/posterior uveitis, retinal vasculitis)
- Skin lesions (erythema nodosum, papulopustular lesions, acneiform nodules)
- Positive pathergy test
The ICBD criteria are now preferred in clinical practice due to higher sensitivity, particularly for recognising vascular and neurological disease manifestations. The ISG criteria remain important for research and clinical trials.
Laboratory Investigations
Non-Specific Inflammatory Markers:
- ESR/CRP: Elevated during disease flares; useful for monitoring activity but non-specific
- Full blood count: Leukocytosis, thrombocytosis during active disease; anaemia of chronic disease
- Acute phase reactants: Ferritin, fibrinogen elevated [63]
Immunological Tests:
-
HLA-B51 typing: Present in 50-70% of endemic populations, 10-30% in Western populations
- Odds ratio for disease 5.78-6.7
- NOT diagnostic (also present in healthy controls)
- May support diagnosis in ambiguous cases
- Not routinely recommended for diagnosis [24,25]
-
Autoantibodies: Typically negative (unlike SLE, GPA)
- ANA, ANCA, anti-dsDNA usually negative
- Positive autoantibodies should prompt consideration of alternative diagnoses
Pathergy Test [15,40]:
- Technique:
- 20-gauge sterile needle
- Intradermal insertion to 5mm depth
- Forearm volar surface (clean with alcohol, dry completely)
- Four puncture sites typically used
- Read at 24-48 hours
- Positive result: Erythematous papule or pustule > 2mm diameter at puncture site
- Sensitivity: 50-70% in Turkish/Middle Eastern populations; 10-20% in Western populations
- Specificity: 90-98%
- Important: Clean technique essential; contamination causes false positives
Exclusion of Mimics:
- HSV PCR/culture: Exclude herpetic genital ulcers
- Syphilis serology (VDRL/TPPA): Exclude syphilitic ulcers
- HIV testing: Particularly with oral ulcers and systemic symptoms
- Interferon-gamma release assay (IGRA): Exclude tuberculosis if considering immunosuppression
Organ-Specific Investigations
Ophthalmological Assessment:
- Slit-lamp examination: Evaluate anterior chamber, hypopyon, keratic precipitates
- Fundoscopy (dilated): Assess retinal vasculitis, vitritis, macular oedema, optic disc changes
- Fluorescein angiography: Demonstrate retinal vessel leakage, occlusion, neovascularisation
- Optical coherence tomography (OCT): Quantify macular oedema, monitor treatment response
- Baseline and regular monitoring essential for all BD patients, even without ocular symptoms [12,64]
Neurological Assessment (if neurological symptoms):
- MRI brain/spine:
- "Parenchymal disease: Brainstem, basal ganglia, hemispheric white matter lesions"
- T2/FLAIR hyperintensity, variable enhancement
- Atrophy in chronic disease
- MR/CT venography: Cerebral venous sinus thrombosis
- CSF analysis: Lymphocytic pleocytosis (10-300 cells/μL), elevated protein (50-150 mg/dL), normal glucose
- EEG: If seizures present [46,47]
Vascular Imaging (if vascular symptoms):
- Doppler ultrasound: Initial assessment of suspected DVT, superficial thrombophlebitis
- CT angiography:
- Pulmonary artery aneurysms (life-threatening)
- Aortic aneurysms, peripheral arterial disease
- Superior sensitivity for arterial lesions
- MR angiography/venography:
- Venous thrombosis (IVC, hepatic veins - Budd-Chiari)
- Non-invasive alternative to CT
- Conventional angiography: Rarely needed; reserved for intervention planning [43,44,45]
Gastrointestinal Assessment (if GI symptoms):
- Oesophagogastroduodenoscopy (OGD): Upper GI ulceration
- Colonoscopy with ileoscopy:
- Ileocaecal region most commonly affected
- Discrete, deep, round/oval ulcers with sharp margins
- Fewer in number than Crohn's disease
- Small bowel imaging (MR enterography, capsule endoscopy): If small bowel involvement suspected
- Histology: Vasculitis, neutrophilic infiltration; absence of granulomas (unlike Crohn's) [48,49]
Monitoring Investigations
Regular Monitoring During Treatment:
- FBC, renal function, liver function (for azathioprine, methotrexate, cyclosporine toxicity)
- Blood pressure (cyclosporine)
- Lipid profile (corticosteroids)
- Bone density (DEXA scan if long-term corticosteroids)
- Thiopurine methyltransferase (TPMT) activity before starting azathioprine
- Regular ophthalmology review (6-monthly minimum, more frequent if active ocular disease) [65]
7. Management
Management of Behçet's disease is tailored to the specific organ systems involved and severity of manifestations. A multidisciplinary approach involving rheumatology, ophthalmology, neurology, dermatology, and vascular surgery is often required.
General Principles
- Organ-based treatment: Mucocutaneous disease treated less aggressively than sight-threatening ocular or life-threatening vascular/neurological disease
- Early aggressive therapy: For organ-threatening disease to prevent irreversible damage
- Multidisciplinary approach: Coordinate care across specialties
- Patient education: Disease course, medication adherence, recognition of complications
- Regular monitoring: For disease activity and treatment toxicity [5,6]
Mucocutaneous Manifestations
Oral and Genital Ulcers:
First-line:
-
Colchicine 0.5-1.5mg daily:
- Reduces frequency and severity of oral and genital ulcers
- RCT evidence for efficacy
- Also benefits erythema nodosum and arthralgia
- "Main side effect: diarrhoea (dose-dependent) [66,67]"
-
Topical corticosteroids:
- Triamcinolone acetonide 0.1% oral paste
- Betamethasone 0.05% cream for genital ulcers
- Reduces pain and healing time
- Minimal systemic absorption [68]
Second-line (if inadequate response):
-
Azathioprine 2-2.5mg/kg/day:
- Effective for recurrent mucocutaneous lesions
- Check TPMT before starting
- Monitor FBC, LFTs monthly initially
- RCT evidence for efficacy [69]
-
Apremilast 30mg twice daily:
- Oral phosphodiesterase-4 inhibitor
- "RELIEF trial: significant reduction in oral ulcers vs placebo"
- "Well tolerated; common side effects: diarrhoea, nausea, headache"
- Expensive; may be limited by funding [70]
-
Dapsone 100mg daily:
- Mucocutaneous lesions, especially if pustular skin lesions
- Check G6PD deficiency before starting
- Monitor for haemolysis, methaemoglobinaemia [71]
Refractory cases:
- Thalidomide 50-100mg daily:
- Highly effective for oral/genital ulcers
- Contraindicated in pregnancy (teratogenic)
- Peripheral neuropathy risk (dose and duration-dependent)
- Requires pregnancy prevention programme [72]
Erythema Nodosum and Skin Lesions:
- Colchicine first-line
- Systemic corticosteroids for severe/acute flares
- Azathioprine for chronic/recurrent disease [73]
Ocular Manifestations
Eye disease requires prompt and aggressive treatment to prevent irreversible visual loss.
Anterior Uveitis (without posterior segment involvement):
- Topical corticosteroids: Prednisolone acetate 1% drops hourly initially
- Mydriatic-cycloplegic: Cyclopentolate 1% or atropine 1% (prevent posterior synechiae)
- Oral corticosteroids: If severe or frequent recurrence
- Azathioprine 2-2.5mg/kg/day: Steroid-sparing agent for recurrent anterior uveitis [74]
Posterior Uveitis, Panuveitis, Retinal Vasculitis (sight-threatening):
Acute treatment:
- High-dose corticosteroids:
- Oral prednisolone 1mg/kg/day OR
- IV methylprednisolone 500-1000mg daily for 3 days (if very severe)
- Taper over 2-3 months based on response [12]
Immunosuppression (essential for most patients):
-
Azathioprine 2-2.5mg/kg/day:
- First-line conventional immunosuppressant
- Reduces uveitis attacks by ~50%
- Takes 6-8 weeks for full effect
- RCT evidence [75]
-
Cyclosporine 3-5mg/kg/day:
- Effective for posterior uveitis
- Faster onset than azathioprine
- "Side effects: nephrotoxicity, hypertension, hypertrichosis, gingival hyperplasia"
- Requires regular BP and renal function monitoring
- Can combine with azathioprine for refractory disease [76]
-
Interferon-alpha-2a 3-6 million IU subcutaneously 3 times weekly:
- Effective for refractory ocular disease
- RCT evidence
- "Side effects: flu-like symptoms, depression, bone marrow suppression"
- Requires monitoring [77]
Biological Therapy (for refractory or very severe ocular disease):
-
Infliximab (anti-TNF):
- "Loading: 5mg/kg at weeks 0, 2, 6, then every 6-8 weeks"
- Superior to conventional immunosuppression for sight-threatening uveitis
- Multiple RCTs demonstrate rapid and sustained response
- Reduces uveitis attacks and improves visual outcomes
- Screen for TB before starting [13,78]
-
Adalimumab (anti-TNF):
- 40mg subcutaneous every 2 weeks (loading 80mg week 0)
- "VISUAL I and II trials: effective for active and inactive uveitis"
- Licensed indication in several countries for BD-related uveitis
- Alternative to infliximab [14,79]
-
Other biologics: Tocilizumab (anti-IL-6), rituximab (anti-CD20), secukinumab (anti-IL-17) reported in case series for refractory cases [80]
EULAR Recommendations for Ocular BD [5]:
- All patients should have baseline ophthalmology assessment
- Acute posterior uveitis/retinal vasculitis: high-dose corticosteroids + azathioprine or anti-TNF
- First-line for sight-threatening disease: azathioprine + systemic corticosteroids
- If azathioprine contraindicated/ineffective: cyclosporine, interferon-alpha, or anti-TNF
- Anti-TNF therapy (infliximab or adalimumab) should be considered early for severe/refractory ocular disease
- Monotherapy with cyclosporine, azathioprine, or methotrexate should be avoided in severe ocular disease
Vascular Manifestations
Venous Thrombosis:
Acute DVT:
- Immunosuppression: Treat underlying inflammation
- High-dose corticosteroids (prednisolone 1mg/kg/day)
- Plus azathioprine 2-2.5mg/kg/day OR cyclophosphamide (if severe)
- Anticoagulation: CONTROVERSIAL
- No high-quality RCT evidence
- "EULAR: immunosuppression is the main treatment; anticoagulation may be used but is not mandatory"
- Risk of bleeding (especially if pulmonary artery aneurysm co-exists)
- "If used: standard therapeutic dose for 3-6 months"
- Low risk of pulmonary embolism (sticky adherent thrombi)
- Consider case-by-case [81,82]
Pulmonary Artery Aneurysm (life-threatening):
- High-dose corticosteroids + cyclophosphamide:
- Cyclophosphamide 600-1000mg/m² IV monthly or 2mg/kg/day oral
- Aggressive immunosuppression essential
- Anti-TNF therapy: If inadequate response or contraindication to cyclophosphamide [83]
- NO anticoagulation: Contraindicated due to high bleeding risk from aneurysm rupture
- Embolisation or surgery: Considered for large aneurysms at high risk of rupture [45]
Arterial Disease (aneurysm, stenosis, occlusion):
- High-dose corticosteroids + cyclophosphamide or anti-TNF
- Surgical intervention requires medical control of inflammation first (anastomotic aneurysm risk if active inflammation)
- Endovascular techniques preferred when feasible [43]
Anticoagulation in BD Venous Thrombosis:
This remains one of the most controversial areas in BD management. Arguments:
Against routine anticoagulation:
- Thrombosis driven by inflammation/vasculitis, not classic thrombophilia
- Low risk of PE (adherent thrombi)
- Bleeding risk, especially with concurrent pulmonary artery aneurysm
- No RCT evidence of benefit
For selective anticoagulation:
- Standard VTE guidelines suggest anticoagulation for DVT
- May reduce extension/recurrence
- Some observational data suggest benefit
Current consensus: Immunosuppression is the cornerstone. Anticoagulation may be considered for large vessel thrombosis without arterial involvement, but is not mandatory. Absolutely contraindicated if pulmonary artery aneurysm present. [81,82]
Neurological Manifestations
Parenchymal Neuro-Behçet's:
Acute presentation:
- High-dose corticosteroids:
- IV methylprednisolone 1000mg daily for 3-7 days
- Followed by oral prednisolone 1mg/kg/day, taper over months
- Immunosuppression:
- Azathioprine 2-2.5mg/kg/day OR
- Methotrexate 15-25mg weekly OR
- Cyclophosphamide for severe/refractory disease
- Anti-TNF therapy: Increasingly used for refractory or severe parenchymal disease
- Infliximab most evidence
- Case series show good response [84,85]
Chronic progressive neuro-Behçet's:
- Methotrexate or azathioprine maintenance
- Anti-TNF if inadequate response
- Avoid cyclosporine (neurotoxic; may worsen CNS disease) [86]
Cerebral Venous Sinus Thrombosis:
- High-dose corticosteroids + immunosuppression (as above)
- Anticoagulation: more accepted for cerebral venous thrombosis than peripheral venous thrombosis
- Therapeutic anticoagulation for 3-6 months typical
- Re-evaluate based on re-imaging [87]
Gastrointestinal Manifestations
Uncomplicated GI Ulcers:
- 5-aminosalicylates (mesalazine): First-line, similar to IBD treatment
- Sulfasalazine 2-4g/day in divided doses
- Corticosteroids: For acute severe disease
- Azathioprine or anti-TNF therapy: For refractory or severe disease [88]
Complicated GI Disease (perforation, fistula, massive haemorrhage):
- Surgical intervention often required
- Medical optimisation pre-operatively if possible
- High risk of complications; specialist gastroenterology/surgery input essential [49]
Musculoskeletal Manifestations
Arthritis/Arthralgia:
- Colchicine 0.5-1.5mg daily: First-line
- NSAIDs: Symptomatic relief (caution with GI involvement)
- Intra-articular corticosteroids: If monoarticular
- Azathioprine: If frequent/severe arthritis not responding to colchicine
- Biologics rarely needed for isolated joint disease [50]
Special Populations
Pregnancy:
- BD can improve, worsen, or remain stable during pregnancy
- Maintain disease control before conception
- Safe medications: colchicine, sulfasalazine, azathioprine, prednisolone
- Avoid: methotrexate (teratogenic), cyclophosphamide (teratogenic), thalidomide (highly teratogenic)
- Anti-TNF: limited data; infliximab and adalimumab may be continued if essential
- Close monitoring by rheumatology and high-risk obstetrics [89]
Elderly:
- Higher risk of medication toxicity (corticosteroids, immunosuppressants)
- Cardiovascular risk with corticosteroids
- Increased infection risk
- Dose adjustment for renal/hepatic impairment
- Regular bone density monitoring; prophylactic bisphosphonates for corticosteroid-induced osteoporosis [90]
8. Complications
| Complication | Frequency | Prevention | Management |
|---|---|---|---|
| Blindness/Severe Visual Loss | 10-25% over 5-10 years if untreated | Early aggressive immunosuppression, regular ophthalmology monitoring | Anti-TNF therapy, visual rehabilitation |
| Neurological Disability | 15-30% of neuro-Behçet's cases develop chronic deficits | Prompt treatment of acute episodes, maintenance immunosuppression | Neurorehabilitation, continued immunosuppression |
| Pulmonary Artery Aneurysm Rupture | 30% mortality if ruptures | Early detection via CT, aggressive immunosuppression | Emergency embolisation/surgery, ICU support |
| Venous Thromboembolism | 15-40% of patients | Immunosuppression, ? anticoagulation | Immunosuppression ± anticoagulation (controversial) |
| Stroke (from arterial occlusion or cerebral venous thrombosis) | 5-10% of patients | Control vascular inflammation, antiplatelet agents | Acute stroke protocol, immunosuppression |
| GI Perforation | 1-5% of GI BD | Early recognition and treatment of GI disease | Emergency surgery, immunosuppression |
| Medication-related: Infection, bone marrow suppression, liver toxicity | Variable by agent | Monitoring, prophylaxis (PJP, vaccinations), patient education | Dose reduction/discontinuation, specific management |
| Corticosteroid-related: Osteoporosis, AVN, diabetes, weight gain | Common with chronic use | Bone protection, lowest effective dose, steroid-sparing agents | Bisphosphonates, diabetes management, AVN surgery if needed |
| Amyloidosis (AA type) | Rare; less than 1% | Control chronic inflammation | Supportive; continue immunosuppression |
9. Prognosis
The clinical course of Behçet's disease is highly variable and characterised by unpredictable exacerbations and remissions.
Overall Mortality:
- 10-year survival > 90% in most cohorts
- Improved significantly over past 3 decades due to better treatments
- Mortality primarily from vascular (pulmonary artery aneurysm rupture, major vessel thrombosis) and neurological complications [10,91]
Morbidity:
- Ocular disease: Major cause of morbidity; blindness in 10-25% at 10 years if inadequately treated
- Neurological disease: Chronic cognitive impairment, motor deficits in 15-30% of neuro-Behçet's patients
- Quality of life: Significantly impaired, particularly during active disease; fatigue, pain, psychological distress common [42,92]
Favourable Prognostic Factors:
- Female sex (less severe disease)
- Older age at onset
- Isolated mucocutaneous disease
- Good response to initial treatment
- Western European/North American ethnicity [93]
Poor Prognostic Factors:
- Male sex (more severe ocular, vascular, neurological disease)
- Young age at onset (less than 25 years)
- Ocular involvement (especially posterior uveitis)
- Neurological involvement
- Vascular involvement (especially arterial)
- HLA-B51 positivity (some studies)
- Middle Eastern/Asian ethnicity [93,94]
Disease Course Over Time:
- Typically most active in first 5-10 years after diagnosis
- Tendency toward gradual improvement/burn-out after 15-20 years in many patients
- Mucocutaneous lesions tend to decrease over time
- Neurological disease may progress chronically despite initial improvement [22,95]
10. Prevention & Screening
Primary Prevention:
- No established primary prevention strategies (aetiology not fully understood)
- Genetic counselling for HLA-B51 positive individuals not currently recommended (low predictive value)
Secondary Prevention:
- Early recognition and treatment of organ-threatening manifestations
- Patient education on red flag symptoms requiring urgent assessment
- Smoking cessation (may worsen vascular disease)
- Cardiovascular risk factor modification (important given corticosteroid use and vascular involvement)
Screening:
- No population-based screening recommended
- Family members of BD patients have slightly increased risk but routine screening not indicated
- All diagnosed BD patients should have:
- Baseline ophthalmology assessment (even if asymptomatic)
- Regular ophthalmology review (at least annually; 6-monthly if ocular disease)
- Vascular imaging if symptoms suggest vascular involvement
- Neurological assessment if CNS symptoms [6]
Infection Screening Before Immunosuppression:
- Tuberculosis screening: IGRA or tuberculin skin test, chest X-ray
- Hepatitis B/C serology
- HIV testing (if risk factors)
- Varicella zoster antibodies (if no clear history of chickenpox)
- Consider PJP prophylaxis if high-dose corticosteroids + other immunosuppression [96]
Vaccination:
- Pneumococcal, annual influenza vaccines recommended
- Avoid live vaccines if on immunosuppression
- Ideally complete vaccinations before starting immunosuppression [97]
11. Key Guidelines
EULAR Recommendations for the Management of Behçet's Syndrome (2018 Update) [5]:
- Organ-based treatment approach
- Colchicine for mucocutaneous lesions
- High-dose corticosteroids + azathioprine or anti-TNF for sight-threatening ocular disease
- Immunosuppression (not anticoagulation) as main treatment for venous thrombosis
- Avoid cyclosporine in neurological disease
- Anti-TNF therapy for refractory major organ involvement
ACR/EULAR Provisional Diagnostic Criteria [11]:
- ICBD criteria recommended for clinical practice (higher sensitivity than ISG)
British Society for Rheumatology (BSR) Guidelines [98]:
- Multidisciplinary management essential
- Early referral to ophthalmology for all patients
- Anti-TNF therapy for refractory disease
12. Examination Focus
Common Exam Questions
Opening Statement for Vivas:
"Behçet's disease is a chronic relapsing systemic vasculitis of unknown aetiology characterised by recurrent oral and genital ulceration, ocular inflammation, and skin lesions. It is most common along the ancient Silk Road from Turkey through the Middle East to Japan, with prevalence up to 370 per 100,000 in Turkey. The disease is associated with HLA-B51 and involves dysregulated innate and adaptive immunity with neutrophil hyperactivity and Th17 polarisation. Diagnosis is clinical using the ICBD or ISG criteria. Management is organ-based, ranging from colchicine for mucocutaneous disease to anti-TNF therapy for sight-threatening ocular or life-threatening vascular manifestations."
Key Viva Points to Memorise:
-
Diagnostic Criteria:
- ISG: Recurrent oral ulcers (≥3 episodes in 12 months) PLUS 2 of: genital ulcers, eye lesions, skin lesions, positive pathergy
- ICBD: Score ≥4 from: oral aphthosis (2 points), genital aphthosis (2), ocular lesions (2), skin lesions (1), vascular (1), pathergy (1), neurological (1)
- ICBD more sensitive (94% vs 85%); ISG more specific
-
HLA-B51 Association:
- Strongest genetic association; OR 5.78-6.7
- Present in 50-70% of endemic populations
- Only accounts for ~20% of genetic risk
- NOT diagnostic (also in healthy controls)
-
Pathergy Test:
- Positive in 50-70% Turkey/Middle East; 10-20% Western populations
- Technique: 20-gauge needle, 5mm intradermal, forearm, read 24-48h
- Positive: erythematous papule/pustule > 2mm
-
Sight-threatening Ocular Disease:
- Posterior uveitis, retinal vasculitis, panuveitis
- Requires urgent systemic immunosuppression
- First-line: corticosteroids + azathioprine OR anti-TNF (infliximab/adalimumab)
- Blindness risk 10-25% at 10 years if inadequately treated
-
Vascular Management - Anticoagulation Debate:
- Immunosuppression is primary treatment for venous thrombosis
- Anticoagulation controversial; may be used but not mandatory
- Absolutely CONTRAINDICATED if pulmonary artery aneurysm present (bleeding risk)
- Low risk of PE (adherent thrombi)
-
Pulmonary Artery Aneurysm:
- Life-threatening; 30% mortality if ruptures
- Presents: haemoptysis, chest pain
- Treatment: high-dose steroids + cyclophosphamide or anti-TNF
- NO anticoagulation
-
Neuro-Behçet's Classification:
- Parenchymal (70-80%): brainstem, meningoencephalitis, cognitive impairment
- Non-parenchymal (20-30%): cerebral venous sinus thrombosis
- Avoid cyclosporine (neurotoxic)
-
Anti-TNF Evidence:
- Infliximab and adalimumab superior to conventional therapy for refractory ocular disease
- VISUAL trials: adalimumab effective for active and inactive uveitis
- Increasingly used for neuro-Behçet's and vascular disease
Common Mistakes
❌ Mistakes that fail candidates:
-
Diagnostic Errors:
- Diagnosing BD based on oral ulcers alone (must have systemic features)
- Not knowing the ISG mandatory criterion (recurrent oral ulcers ≥3 episodes in 12 months)
- Thinking HLA-B51 is diagnostic (it's an association, not a diagnostic test)
-
Management Errors:
- Using cyclosporine for neuro-Behçet's (neurotoxic; contraindicated)
- Anticoagulating patients with pulmonary artery aneurysm (major bleeding risk)
- Failing to urgently refer posterior uveitis for systemic immunosuppression
- Not screening for TB before starting immunosuppression
-
Knowledge Gaps:
- Not knowing difference between ISG and ICBD criteria
- Unable to explain pathergy test technique
- Not recognising that immunosuppression (not anticoagulation) is primary treatment for venous thrombosis in BD
- Thinking BD is primarily an autoimmune disease (it's autoinflammatory with vasculitis)
Model Answers
Q: A 30-year-old Turkish man presents with recurrent painful oral ulcers, genital ulcers, and uveitis. What is your approach to diagnosis and management?
A: "This presentation is highly suggestive of Behçet's disease given the triad of recurrent oral ulcers, genital ulcers, and uveitis in a young man from an endemic region.
For diagnosis, I would:
- Take detailed history confirming recurrence pattern (oral ulcers ≥3 episodes in 12 months for ISG criteria)
- Examine for skin lesions (erythema nodosum, papulopustular lesions)
- Perform pathergy testing (though sensitivity varies by population)
- Apply clinical diagnostic criteria - this patient already meets ISG criteria (recurrent oral ulcers + genital ulcers + eye disease = 3/4 criteria) and ICBD criteria (2+2+2 = 6 points)
- Arrange urgent ophthalmology assessment to characterise uveitis (anterior vs posterior segment)
- Send inflammatory markers, FBC, HLA-B51 typing (supportive but not diagnostic), and exclusion tests (HSV PCR/culture for genital ulcers, syphilis serology, autoantibodies to exclude SLE/vasculitis)
For management:
- If anterior uveitis only: topical corticosteroids, mydriatic, plus colchicine for mucocutaneous lesions and systemic azathioprine as steroid-sparing agent
- If posterior uveitis/retinal vasculitis (sight-threatening): urgent high-dose systemic corticosteroids (prednisolone 1mg/kg) plus azathioprine 2-2.5mg/kg OR anti-TNF therapy (infliximab/adalimumab) per EULAR guidelines
- Multidisciplinary care with rheumatology and ophthalmology
- Patient education on red flags requiring urgent review
- Before starting immunosuppression: screen for TB, hepatitis B/C, check TPMT if using azathioprine
The goal is to prevent irreversible visual loss through early aggressive immunosuppression of sight-threatening disease."
Q: What is the role of anticoagulation in Behçet's disease with deep vein thrombosis?
A: "This is a controversial area in Behçet's management. The key principle is that venous thrombosis in BD is driven by inflammatory vasculitis rather than classic hypercoagulability, which has important treatment implications.
Current Evidence and Guidelines:
- EULAR 2018 guidelines state that immunosuppression is the main treatment for BD-related venous thrombosis
- Anticoagulation may be used but is not mandatory
- There are no high-quality RCTs comparing anticoagulation vs no anticoagulation in BD
Arguments Against Routine Anticoagulation:
- Thrombosis mechanism is inflammatory vasculitis, not thrombophilia
- Thrombi are characteristically adherent and sticky with very low risk of pulmonary embolism
- Bleeding risk, particularly if concurrent pulmonary artery aneurysm (present in up to 10% of BD patients with thrombosis)
- No RCT evidence of benefit
Arguments For Selective Use:
- Large vessel thrombosis may benefit
- Standard VTE guidelines recommend anticoagulation
- Some observational data suggest reduced recurrence
My Approach:
- Primary treatment: high-dose corticosteroids (prednisolone 1mg/kg) + azathioprine or cyclophosphamide to treat underlying vasculitis
- Essential first step: imaging to exclude pulmonary artery aneurysm (CT pulmonary angiography)
- If pulmonary artery aneurysm present: absolutely NO anticoagulation due to bleeding risk
- If no arterial involvement and large proximal DVT: may consider therapeutic anticoagulation for 3-6 months alongside immunosuppression, but this is not mandatory
- Close monitoring and individualised decision-making
The critical point is that immunosuppression is the cornerstone of treatment, and anticoagulation should never be given without first excluding pulmonary artery aneurysm."
References
-
Yazici H, Seyahi E, Hatemi G, Yazici Y. Behçet syndrome: a contemporary view. Nat Rev Rheumatol. 2018;14(2):107-119. doi:10.1038/nrrheum.2017.208
-
Sakane T, Takeno M, Suzuki N, Inaba G. Behçet's disease. N Engl J Med. 1999;341(17):1284-1291. doi:10.1056/NEJM199910213411707
-
Criteria for diagnosis of Behçet's disease. International Study Group for Behçet's Disease. Lancet. 1990;335(8697):1078-1080. PMID: 1970380
-
Davatchi F, Assaad-Khalil S, Calamia KT, et al. The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338-347. doi:10.1111/jdv.12107
-
Hatemi G, Christensen R, Bang D, et al. 2018 update of the EULAR recommendations for the management of Behçet's syndrome. Ann Rheum Dis. 2018;77(6):808-818. doi:10.1136/annrheumdis-2018-213225
-
Yazici Y, Yurdakul S, Yazici H. Behçet's syndrome. Curr Rheumatol Rep. 2010;12(6):429-435. doi:10.1007/s11926-010-0132-z
-
Mahr A, Maldini C. Epidemiology of Behçet's disease. Rev Med Interne. 2014;35(2):81-89. doi:10.1016/j.revmed.2013.12.005
-
Zeidan MJ, Saadoun D, Garrido M, Klatzmann D, Six A, Cacoub P. Behçet's disease physiopathology: a contemporary review. Auto Immun Highlights. 2016;7(1):4. doi:10.1007/s13317-016-0074-1
-
Tursen U. Pathophysiology of the Behçet's disease. Patholog Res Int. 2012;2012:493015. doi:10.1155/2012/493015
-
Kural-Seyahi E, Fresko I, Seyahi N, et al. The long-term mortality and morbidity of Behçet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore). 2003;82(1):60-76. doi:10.1097/00005792-200301000-00006
-
International Team for the Revision of the International Criteria for Behçet's Disease (ITR-ICBD). The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338-347. doi:10.1111/jdv.12107
-
Tugal-Tutkun I. Behçet's uveitis. Middle East Afr J Ophthalmol. 2009;16(4):219-224. doi:10.4103/0974-9233.58425
-
Hatemi G, Melikoglu M, Tunc R, et al. Apremilast for Behçet's syndrome--a phase 2, placebo-controlled study. N Engl J Med. 2015;372(16):1510-1518. doi:10.1056/NEJMoa1408684
-
Jaffe GJ, Dick AD, Brézin AP, et al. Adalimumab in patients with active noninfectious uveitis. N Engl J Med. 2016;375(10):932-943. doi:10.1056/NEJMoa1509852
-
Fresko I, Hamuryudan V, Demir M, et al. Standardization of the pathergy test in Behçet's disease: microtrauma at 48 h. Clin Exp Rheumatol. 2004;22(4 Suppl 34):S45. PMID: 15515785
-
Davatchi F, Shahram F, Chams-Davatchi C, et al. Behcet's disease: from East to West. Clin Rheumatol. 2010;29(8):823-833. doi:10.1007/s10067-010-1430-6
-
Ohno S, Ohguchi M, Hirose S, Matsuda H, Wakisaka A, Aizawa M. Close association of HLA-Bw51 with Behçet's disease. Arch Ophthalmol. 1982;100(9):1455-1458. doi:10.1001/archopht.1982.01030040433013
-
Bang D, Lee JH, Lee ES, et al. Epidemiologic and clinical survey of Behçet's disease in Korea: the first multicenter study. J Korean Med Sci. 2001;16(5):615-618. doi:10.3346/jkms.2001.16.5.615
-
Koné-Paut I, Shahram F, Darce-Bello M, et al. Consensus classification criteria for paediatric Behçet's disease from a prospective observational cohort: PEDBD. Ann Rheum Dis. 2016;75(6):958-964. doi:10.1136/annrheumdis-2015-208491
-
Borlu M, Ukşal U, Ferahbaş A, Evereklioglu C. Clinical features of Behçet's disease in children. Int J Dermatol. 2006;45(6):713-716. doi:10.1111/j.1365-4632.2006.02877.x
-
de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behçet's disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. 2009;61(10):1287-1296. doi:10.1002/art.24642
-
Takeuchi M, Kastner DL, Remmers EF. The immunogenetics of Behçet's disease: A comprehensive review. J Autoimmun. 2015;64:137-148. doi:10.1016/j.jaut.2015.08.013
-
Yoshida A, Kawashima H, Motoyama Y, et al. Comparison of patients with Behçet's disease in the 1980s and 1990s. Ophthalmology. 2004;111(4):810-815. doi:10.1016/j.ophtha.2003.07.018
-
Hughes T, Coit P, Adler A, et al. Identification of multiple independent susceptibility loci in the HLA region in Behçet's disease. Nat Genet. 2013;45(3):319-324. doi:10.1038/ng.2551
-
Verity DH, Marr JE, Ohno S, Wallace GR, Stanford MR. Behçet's disease, the Silk Road and HLA-B51: historical and geographical perspectives. Tissue Antigens. 1999;54(3):213-220. doi:10.1034/j.1399-0039.1999.540301.x
-
Mendes D, Correia M, Barbedo M, et al. Behçet's disease--a contemporary review. J Autoimmun. 2009;32(3-4):178-188. doi:10.1016/j.jaut.2009.02.011
-
Kirino Y, Bertsias G, Ishigatsubo Y, et al. Genome-wide association analysis identifies new susceptibility loci for Behçet's disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. 2013;45(2):202-207. doi:10.1038/ng.2520
-
Fei Y, Webb R, Cobb BL, Direskeneli H, Saruhan-Direskeneli G, Sawalha AH. Identification of novel genetic susceptibility loci for Behçet's disease using a genome-wide association study. Arthritis Res Ther. 2009;11(3):R66. doi:10.1186/ar2695
-
Kaneko F, Oyama N, Nishibu A. Streptococcal infection in the pathogenesis of Behçet's disease and clinical effects of minocycline on the disease symptoms. Yonsei Med J. 1997;38(6):444-454. doi:10.3349/ymj.1997.38.6.444
-
Consolandi C, Turroni S, Emmi G, et al. Behçet's syndrome patients exhibit specific microbiome signature. Autoimmun Rev. 2015;14(4):269-276. doi:10.1016/j.autrev.2014.11.009
-
Eksioglu-Demiralp E, Direskeneli H, Kibaroglu A, Yavuz S, Ergun T, Akoglu T. Neutrophil activation in Behçet's disease. Clin Exp Rheumatol. 2001;19(5 Suppl 24):S19-24. PMID: 11760395
-
Sohn S. Immunopathogenesis of Behçet's disease: the role of innate immunity. Adv Exp Med Biol. 2003;528:49-54. doi:10.1007/0-306-48382-3_10
-
Hamzaoui K, Hamzaoui A, Guemira F, Bessioud M, Hamzaoui M, Ayed K. Cytokine profile in Behçet's disease patients. Relationship with disease activity. Scand J Rheumatol. 2002;31(4):205-210. doi:10.1080/030097402320318378
-
Geri G, Terrier B, Rosenzwajg M, et al. Critical role of IL-21 in modulating TH17 and regulatory T cells in Behçet disease. J Allergy Clin Immunol. 2011;128(3):655-664. doi:10.1016/j.jaci.2011.05.029
-
Bank I, Duvdevani M, Livneh A. Expansion of gammadelta T-cells in Behçet's disease: role of disease activity and microbial flora in oral ulcers. J Lab Clin Med. 2003;141(1):33-40. doi:10.1067/mlc.2003.8
-
Chambers JC, Haskard DO, Kooner JS. Vascular endothelial function and oxidative stress mechanisms in patients with Behçet's syndrome. J Am Coll Cardiol. 2001;37(2):517-520. doi:10.1016/s0735-1097(00)01137-2
-
Direskeneli H. Autoimmunity vs autoinflammation in Behcet's disease: do we oversimplify a complex disorder? Rheumatology (Oxford). 2006;45(12):1461-1465. doi:10.1093/rheumatology/kel329
-
Alpsoy E. Behçet's disease: A comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. J Dermatol. 2016;43(6):620-632. doi:10.1111/1346-8138.13381
-
Fresko I, Yazıcı H, Bayramiçlı M, Yurdakul S, Mat C. Effect of surgical cleaning of the skin on the pathergy phenomenon in Behçet's syndrome. Ann Rheum Dis. 1993;52(8):619-620. doi:10.1136/ard.52.8.619
-
Dilşen N, Konice M, Aral O, Ocal L, Inanç M, Gül A. Comparative study of the skin pathergy test with blunt and sharp needles in Behçet's disease: confirmed specific diagnostic value. Rheumatol Int. 1993;13(2):75-77. doi:10.1007/BF00307734
-
Khairallah M, Accorinti M, Muccioli C, et al. Epidemiology of Behçet disease. Ocul Immunol Inflamm. 2012;20(5):324-335. doi:10.3109/09273948.2012.723112
-
Takeuchi M, Hokama H, Tsukahara R, et al. Risk and prognostic factors of poor visual outcome in Behçet's disease with ocular involvement. Graefes Arch Clin Exp Ophthalmol. 2005;243(11):1147-1152. doi:10.1007/s00417-005-1135-z
-
Seyahi E. Behçet's disease: How to diagnose and treat vascular involvement. Best Pract Res Clin Rheumatol. 2016;30(2):279-295. doi:10.1016/j.berh.2016.08.002
-
Ames PR, Steuer A, Pap A, Denman AM. Thrombosis in Behçet's disease: a retrospective survey from a single UK centre. Rheumatology (Oxford). 2001;40(6):652-655. doi:10.1093/rheumatology/40.6.652
-
Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine (Baltimore). 2012;91(1):35-48. doi:10.1097/MD.0b013e3182428f49
-
Akman-Demir G, Serdaroglu P, Tasçi B. Clinical patterns of neurological involvement in Behçet's disease: evaluation of 200 patients. The Neuro-Behçet Study Group. Brain. 1999;122 ( Pt 11):2171-2182. doi:10.1093/brain/122.11.2171
-
Borhani Haghighi A, Pourmand R, Nikseresht AR. Neuro-Behçet disease. A review. Neurologist. 2005;11(2):80-89. doi:10.1097/01.nrl.0000156203.96107.54
-
Cheon JH, Kim WH. An update on the diagnosis, treatment, and prognosis of intestinal Behçet's disease. Curr Opin Rheumatol. 2015;27(1):24-31. doi:10.1097/BOR.0000000000000125
-
Bayraktar Y, Ozaslan E, Van Thiel DH. Gastrointestinal manifestations of Behçet's disease. J Clin Gastroenterol. 2000;30(2):144-154. doi:10.1097/00004836-200003000-00006
-
Yurdakul S, Yazıcı H, Tüzün Y, et al. The arthritis of Behcet's disease: a prospective study. Ann Rheum Dis. 1983;42(5):505-515. doi:10.1136/ard.42.5.505
-
Geri G, Wechsler B, Thi Huong du L, et al. Spectrum of cardiac lesions in Behçet disease: a series of 52 patients and review of the literature. Medicine (Baltimore). 2012;91(1):25-34. doi:10.1097/MD.0b013e3182428f5f
-
Akpolat T, Koç Y, Yeniay I, et al. Familial Mediterranean fever and Henoch-Schönlein purpura in a patient with Behçet's disease and amyloidosis. Nephrol Dial Transplant. 1998;13(9):2431-2433. doi:10.1093/ndt/13.9.2431
-
Park JH, Han MC, Bettmann MA. Arterial manifestations of Behçet disease. AJR Am J Roentgenol. 1984;143(4):821-825. doi:10.2214/ajr.143.4.821
-
Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 1998;9(3):306-321. doi:10.1177/10454411980090030401
-
Bontems S, Vanhooteghem O, Theate I, de la Brassinne M. Genital herpes simplex infection sometimes mimics Behçet's disease. Rev Med Liege. 2009;64(1):5-9. PMID: 19271662
-
Hirohata S, Kikuchi H. Behçet's disease and systemic lupus erythematosus: a comparison. Lupus. 2018;27(8):1321-1329. doi:10.1177/0961203318776104
-
Agrawal R, Xin W, Keane PA, Crama N, Pavesio C. Tuberculosis: An update on clinical presentation and diagnosis in uveitis. Ocul Immunol Inflamm. 2011;19(5):294-300. doi:10.3109/09273948.2011.608431
-
Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11. doi:10.1002/art.37715
-
Hannu T. Reactive arthritis. Best Pract Res Clin Rheumatol. 2011;25(3):347-357. doi:10.1016/j.berh.2011.01.018
-
Firestein GS, Gruber HE, Weisman MH, et al. Mouth and genital ulcers with inflamed cartilage: MAGIC syndrome. Five patients with features of relapsing polychondritis and Behçet's disease. Am J Med. 1985;79(1):65-72. doi:10.1016/0002-9343(85)90526-1
-
Cohen PR. Sweet's syndrome--a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi:10.1186/1750-1172-2-34
-
International Study Group for Behçet's Disease. Criteria for diagnosis of Behçet's disease. Lancet. 1990;335(8697):1078-1080. PMID: 1970380
-
Erkan F, Gül A, Tasali E. Pulmonary manifestations of Behçet's disease. Thorax. 2001;56(7):572-578. doi:10.1136/thorax.56.7.572
-
Lightman S, Taylor SR, Bunce C, et al. Pegylated interferon-α-2b reduces corticosteroid requirement in patients with Behçet's disease with upregulation of circulating regulatory T cells and reduction of Th17. Ann Rheum Dis. 2015;74(6):1138-1144. doi:10.1136/annrheumdis-2014-205571
-
Sharquie KE, Najim RA, Abu-Raghif AR. Dapsone in Behçet's disease: a double-blind, placebo-controlled, cross-over study. J Dermatol. 2002;29(5):267-279. doi:10.1111/j.1346-8138.2002.tb00264.x
-
Yurdakul S, Mat C, Tüzün Y, et al. A double-blind trial of colchicine in Behçet's syndrome. Arthritis Rheum. 2001;44(11):2686-2692. doi:10.1002/1529-0131(200111)44:11less than 2686::aid-art448> 3.0.co;2-h
-
Davatchi F, Sadeghi Abdollahi B, Tehrani Banihashemi A, et al. Colchicine versus placebo in Behçet's disease: randomized, double-blind, controlled crossover trial. Mod Rheumatol. 2009;19(5):542-549. doi:10.1007/s10165-009-0200-2
-
Alpsoy E, Er H, Durusoy C, Yilmaz E. The use of sucralfate suspension in the treatment of oral and genital ulceration of Behçet disease: a randomized, placebo-controlled, double-blind study. Arch Dermatol. 1999;135(5):529-532. doi:10.1001/archderm.135.5.529
-
Yazici H, Pazarli H, Barnes CG, et al. A controlled trial of azathioprine in Behçet's syndrome. N Engl J Med. 1990;322(5):281-285. doi:10.1056/NEJM199002013220501
-
Hatemi G, Mahr A, Ishigatsubo Y, et al. Trial of Apremilast for Oral Ulcers in Behçet's Syndrome. N Engl J Med. 2019;381(20):1918-1928. doi:10.1056/NEJMoa1816594
-
Sharquie KE, Najim RA, Abu-Raghif AR. Dapsone in Behçet's disease: a double-blind, placebo-controlled, cross-over study. J Dermatol. 2002;29(5):267-279. doi:10.1111/j.1346-8138.2002.tb00264.x
-
Hamuryudan V, Mat C, Saip S, et al. Thalidomide in the treatment of the mucocutaneous lesions of the Behçet syndrome. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1998;128(6):443-450. doi:10.7326/0003-4819-128-6-199803150-00004
-
Mat MC, Goksugur N, Engin B, Yurdakul S, Yazici H. The frequency of scarring after genital ulcers in Behçet's syndrome: a prospective study. Int J Dermatol. 2006;45(5):554-556. doi:10.1111/j.1365-4632.2005.02558.x
-
BenEzra D, Cohen E, Chajek T, et al. Evaluation of conventional therapy versus cyclosporine A in Behçet's syndrome. Transplant Proc. 1988;20(3 Suppl 4):136-143. PMID: 3291272
-
Yazici H, Pazarli H, Barnes CG, et al. A controlled trial of azathioprine in Behçet's syndrome. N Engl J Med. 1990;322(5):281-285. doi:10.1056/NEJM199002013220501
-
Masuda K, Nakajima A, Urayama A, Nakae K, Kogure M, Inaba G. Double-masked trial of cyclosporin versus colchicine and long-term open study of cyclosporin in Behçet's disease. Lancet. 1989;1(8647):1093-1096. doi:10.1016/s0140-6736(89)92384-1
-
Kötter I, Günaydin I, Zierhut M, Stübiger N. The use of interferon alpha in Behçet disease: review of the literature. Semin Arthritis Rheum. 2004;33(5):320-335. doi:10.1016/j.semarthrit.2003.09.010
-
Ohno S, Nakamura S, Hori S, et al. Efficacy, safety, and pharmacokinetics of multiple administration of infliximab in Behçet's disease with refractory uveoretinitis. J Rheumatol. 2004;31(7):1362-1368. PMID: 15229958
-
Fabiani C, Vitale A, Emmi G, et al. Efficacy and safety of adalimumab in Behçet's disease-related uveitis: a multicenter retrospective observational study. Clin Rheumatol. 2017;36(1):183-189. doi:10.1007/s10067-016-3480-x
-
Fabiani C, Sota J, Vitale A, et al. Tocilizumab in patients with Behçet's disease: A case series and a systematic literature review. Clin Exp Rheumatol. 2017;35 Suppl 108(6):148-154. PMID: 29185973
-
Desbois AC, Wechsler B, Resche-Rigon M, et al. Immunosuppressants reduce venous thrombosis relapse in Behçet's disease. Arthritis Rheum. 2012;64(8):2753-2760. doi:10.1002/art.34450
-
Seyahi E, Yazici H. Anticoagulation in Behçet's disease. Lupus. 2015;24(7):761-767. doi:10.1177/0961203314567751
-
Hamuryudan V, Er T, Seyahi E, et al. Pulmonary artery aneurysms in Behçet syndrome. Am J Med. 2004;117(11):867-870. doi:10.1016/j.amjmed.2004.05.027
-
Shapiro LS, Farrell J, Borhani Haghighi A. Tocilizumab treatment for neuro-Behçet's disease, the first report. Clin Neurol Neurosurg. 2012;114(3):297-298. doi:10.1016/j.clineuro.2011.10.024
-
Fasano A, D'Agostino M, Caldarola G, et al. Infliximab monotherapy in neuro-Behçet's disease: four year follow-up in a long-standing case resistant to conventional therapies. J Neuroimmunol. 2011;239(1-2):105-107. doi:10.1016/j.jneuroim.2011.08.018
-
Siva A, Kantarci OH, Saip S, et al. Behçet's disease: diagnostic and prognostic aspects of neurological involvement. J Neurol. 2001;248(2):95-103. doi:10.1007/s004150170270
-
Saadoun D, Wechsler B, Resche-Rigon M, et al. Cerebral venous thrombosis in Behçet's disease. Arthritis Rheum. 2009;61(4):518-526. doi:10.1002/art.24393
-
Ebert EC. Gastrointestinal manifestations of Behçet's disease. Dig Dis Sci. 2009;54(2):201-207. doi:10.1007/s10620-008-0337-4
-
Noel N, Wechsler B, Nizard J, et al. Behçet's disease and pregnancy. Arthritis Rheum. 2013;65(9):2450-2456. doi:10.1002/art.38052
-
Pipitone N, Olivieri I, Padula A, Boiardi L, Salvarani C. Behçet's syndrome in the elderly. Drugs Aging. 2007;24(8):691-694. doi:10.2165/00002512-200724080-00006
-
Alibaz-Oner F, Karadeniz A, Ylmaz S, et al. Behçet disease with vascular involvement: effects of different therapeutic regimens on the incidence of new relapses. Medicine (Baltimore). 2015;94(6):e494. doi:10.1097/MD.0000000000000494
-
Uğuz F, Dursun R, Kaya N, Cilli AS. Quality of life in patients with Behçet's disease: the impact of major depression. Gen Hosp Psychiatry. 2007;29(1):21-24. doi:10.1016/j.genhosppsych.2006.10.001
-
Krause I, Weinberger A. Behçet's disease. Curr Opin Rheumatol. 2008;20(1):82-87. doi:10.1097/BOR.0b013e3282f1516b
-
Monastero RN, Kiran RP. The long-term prognosis of Behçet's disease. Cleve Clin J Med. 2003;70(12):1060-1062. doi:10.3949/ccjm.70.12.1060
-
Kural-Seyahi E, Fresko I, Seyahi N, et al. The long-term mortality and morbidity of Behçet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore). 2003;82(1):60-76. doi:10.1097/00005792-200301000-00006
-
Hamuryudan V, Sonsuz A, Mat C, et al. Interferon alfa combined with conventional therapy for Behçet's disease: a randomized trial. Clin Exp Rheumatol. 2007;25(4 Suppl 45):S63-68. PMID: 17949554
-
van Assen S, Agmon-Levin N, Elkayam O, et al. EULAR recommendations for vaccination in adult patients with autoimmune inflammatory rheumatic diseases. Ann Rheum Dis. 2011;70(3):414-422. doi:10.1136/ard.2010.137216
-
Davies PG, Fordham JN, Kirwan JR, Barnes CG, Dinning WJ. The pathergy test and Behçet's syndrome in Britain. Ann Rheum Dis. 1984;43(1):70-73. doi:10.1136/ard.43.1.70
Last Updated: 2026-01-05
Learning map
Use these linked topics to study the concept in sequence and compare related presentations.
Prerequisites
Start here if you need the foundation before this topic.
- Systemic Vasculitis Overview
Differentials
Competing diagnoses and look-alikes to compare.
- Herpes Simplex Virus Infection
- Recurrent Aphthous Stomatitis
- Crohn's Disease
- Systemic Lupus Erythematosus
- Sarcoidosis