Paeds · genetics-dysmorphology-and-metabolism
Prenatal diagnosis and reproductive genetics
Also known as Prenatal genetic diagnosis · Antenatal genetic testing · Chorionic villus sampling · Amniocentesis · Chromosomal microarray · Prenatal exome sequencing · Preimplantation genetic testing · Carrier screening · Non-invasive prenatal diagnosis · Reproductive genetic counselling
Fellowship guide to prenatal diagnosis and reproductive genetics: screening versus diagnostic genetic testing, first-trimester combined screening and cell-free DNA, chorionic villus sampling and amniocentesis with contemporary procedure-related risk, chromosomal microarray versus karyotype, prenatal exome and genome sequencing, variant interpretation, Mendelian and empirical recurrence risk, expanded carrier screening, preimplantation genetic testing, non-invasive prenatal diagnosis, multidisciplinary coordination, and regional programme differences.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Prenatal diagnosis and reproductive genetics is the set of tests and counselling conversations that allow families and clinical teams to know the genetic status of a fetus before or at birth. The field spans carrier screening before pregnancy, aneuploidy and structural screening during pregnancy, invasive diagnostic procedures, molecular genetic tests of escalating resolution, and reproductive technologies such as preimplantation genetic testing. The paediatrician's role is to participate in antenatal multidisciplinary discussion, translate a confirmed genetic result into a postnatal plan, and own the medical-home function through birth and beyond. [6] [9]
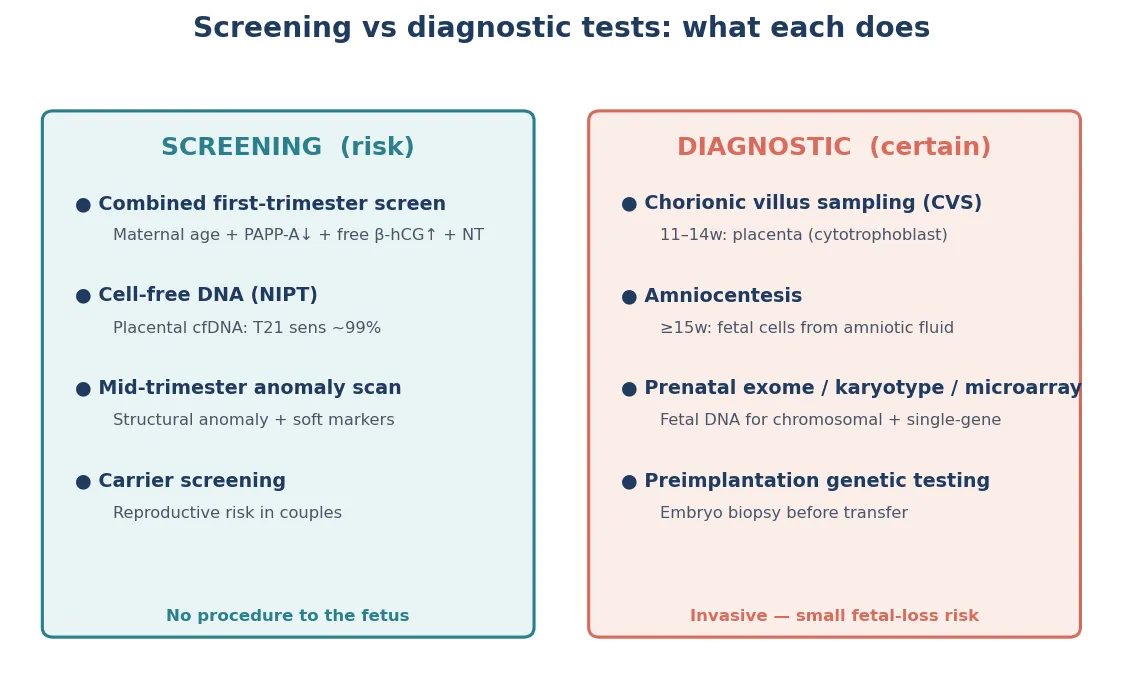
The single most important distinction in this topic is screening versus diagnosis. Screening estimates a probability. Diagnostic testing confirms or excludes a condition. Cell-free DNA screening, first-trimester combined screening and the mid-trimester scan are screens — they tell you whether the chance of a problem is raised. Chorionic villus sampling, amniocentesis and the downstream molecular tests on those samples are diagnostic — they tell you what the chromosomes or genes actually are. Blurring that line is the most common and most dangerous counselling error in reproductive genetics. [3] [7]
The second key distinction is the resolution ladder of genetic testing. A karyotype sees whole chromosomes under the microscope. A chromosomal microarray detects submicroscopic copy-number variants — deletions and duplications — down to the kilobase level, which is why it is first-tier for structurally abnormal fetuses. Exome sequencing interrogates the coding regions of nearly all genes and finds single-nucleotide variants responsible for Mendelian disease. Genome sequencing reads the entire genome. Each step up the ladder increases diagnostic yield but also increases the chance of finding a variant of uncertain significance, which is why the counselling complexity rises with the resolution. [1] [6] [11]
What you actually do across the reproductive genetics pathway
Assess pre-test probability
Maternal age, family history, consanguinity, prior affected child, parental carrier status, BMI — interpret every result against this baseline.
Offer screening
Aneuploidy screening (combined or cfDNA), structural survey, carrier screening — document the result state, never leave a blank.
Confirm diagnostically
CVS or amniocentesis for genuinely raised risk; choose microarray, exome or targeted testing by the clinical question.
Interpret honestly
Pathogenic, likely pathogenic, VUS, likely benign, benign — state what each means and does not mean.
Close the loop
Multidisciplinary planning, reproductive options counselling, neonatal alert, named owner, postnatal confirmation and surveillance.
Classification
Think first in four genetic-assessment domains, each answering a different question about the fetal genome. [1] [6]
Aneuploidy screening estimates the chance of a whole-chromosome trisomy or sex-chromosome aneuploidy — trisomy 21, 18, 13, and conditions such as Turner and Klinefelter syndromes. Structural sonographic survey looks for physical malformations on the mid-trimester anatomy scan that may trigger diagnostic genetic testing. Diagnostic cytogenetic testing — karyotype, chromosomal microarray — confirms or excludes chromosomal abnormalities at increasing resolution. Molecular diagnostic testing — exome, genome, targeted single-gene analysis — interrogates the DNA sequence itself for monogenic disease. Mixing these up is how clinicians reassure themselves with the wrong test. [1] [9]
Then classify the result state. Low-risk means continue routine care, but residual risk persists. Increased-risk means counsel and offer diagnostic testing — it is not a diagnosis. Inconclusive (a no-call cell-free DNA result, a variant of uncertain significance, or a technically limited sample) is its own state with its own risk, not a pass. Declined means the family chose not to proceed, and it needs documentation and an open door, not coercion. Each state demands a different action, and confusing them is a common exam trap. [3] [7]
Within diagnostic cytogenetics, the resolution ladder matters enormously. Karyotype detects aneuploidies and large rearrangements visible at approximately five to ten megabase resolution. Chromosomal microarray — using array comparative genomic hybridisation or single-nucleotide polymorphism arrays — detects copy-number variants down to approximately 50 to 200 kilobases and is recommended as first-tier for fetuses with structural anomalies because it increases diagnostic yield by approximately six percentage points over karyotype alone. Exome sequencing finds the single-nucleotide variants and small insertions or deletions responsible for Mendelian disease that both karyotype and microarray miss. [1] [6] [11]
Within reproductive genetics, three carrier screening strategies exist. Population-based screening targets specific high-frequency conditions in defined ethnic groups — for example, Tay-Sachs in Ashkenazi Jewish populations or haemoglobinopathies in Mediterranean and South Asian populations. Expanded (pan-ethnic) carrier screening uses a multi-gene panel offered to all couples regardless of ancestry, on the rationale that pan-ethnic screening removes the reliance on self-reported ethnicity that misses admixed and unsuspected carriers. Cascade screening traces relatives of a known carrier or affected individual. Know which programme your jurisdiction uses. [7]
Preimplantation genetic testing is classified by its target. PGT-M (monogenic) tests embryos for a known familial single-gene disorder using linkage analysis and direct mutation detection. PGT-SR (structural rearrangement) tests embryos when a parent carries a balanced translocation or inversion. PGT-A (aneuploidy) screens embryos for chromosomal copy-number changes to prioritise transfer of euploid embryos, though its clinical utility remains debated. These are distinct technologies with distinct indications and limitations. [8]
The common exam trap is treating a screen and a diagnostic test as interchangeable, or treating a variant of uncertain significance as either pathogenic or benign. Both are failures of reasoning. [1] [6]
Screen
Estimates probability
- First-trimester combined, cfDNA, quad serum, anatomy scan
- Output is a risk, not a diagnosis
- A positive result triggers counselling and diagnostic testing
- A no-call is its own state, not low risk
Diagnostic
Confirms or excludes
- CVS, amniocentesis, fetal blood sampling
- Microarray, exome, targeted gene testing
- Carries a small procedure-related risk
- Residual risk may persist for uninterrogated genes
Pathogenic
Confirmed finding
- Genotype correlates with a known phenotype
- Drives prognosis and neonatal planning
- Reproductive recurrence risk is calculable
- Multidisciplinary pathway opens
VUS
Unknown meaning
- Real finding, uncertain clinical significance
- Neither pathogenic nor benign by evidence
- Parental testing and reanalysis over time
- Do not over-call or dismiss
Epidemiology & Risk Factors
Major chromosomal aneuploidies are uncommon at the individual level but common enough at population level to justify universal screening. Trisomy 21 is the single most common survivable autosomal trisomy, and its probability rises steeply with maternal age — from approximately 1 in 1,500 at age 20 to approximately 1 in 100 at age 40. That is why advanced maternal age increases pre-test probability but does not, on its own, define a high-risk pregnancy. The population-level birth prevalence of aneuploidy has shifted as universal screening and earlier diagnosis change the landscape of detected and continued pregnancies. [3] [7]
Risk factors change counselling in two ways. They raise pre-test probability, which changes the meaning of any result, and they raise the chance of a no-call or inconclusive result. Maternal age, prior affected pregnancy, a parental balanced translocation, consanguinity, high body mass index and assisted reproduction all shift the baseline. A family history of a Mendelian condition moves the conversation toward targeted diagnostic testing rather than population aneuploidy screening. Advanced paternal age raises the risk of de novo dominant mutations associated with some conditions, including achondroplasia and other FGFR-related skeletal dysplasias. [7]
Cell-free DNA has a specific failure mode worth knowing. A no-call result — when the laboratory cannot report because fetal fraction is too low — is more likely with high maternal weight, early gestational age at sampling, aneuploidy itself and fetal growth restriction. A no-call is therefore not a neutral outcome. It is a signal that carries its own risk of aneuploidy and placental insufficiency, and it demands an action — repeat, diagnose or surveil — rather than a re-labelling as low-risk. [3] [7]
The carrier frequency of common autosomal recessive conditions justifies expanded carrier screening. Cystic fibrosis affects approximately 1 in 2,500 Northern European births with a carrier frequency near 1 in 25; spinal muscular atrophy has a carrier frequency near 1 in 50; fragile X premutation carrier frequency is approximately 1 in 150000 to 1 in 300 in women. In populations with founder effects and high consanguinity, the aggregate autosomal recessive risk rises substantially. These numbers are why pan-ethnic expanded carrier screening is increasingly offered to all couples before or early in pregnancy. [7]
Access barriers are part of the epidemiology, not an afterthought. Rural families, language-discordant families, those without reliable transport or leave from work, and those in lower socioeconomic groups complete fewer screens and fewer diagnostic pathways. Incomplete pathways are where prenatal diagnosis silently fails, and the resulting postnatal surprises — an unanticipated syndrome, a missed metabolic condition, an unprepared neonatal team — are preventable harms that fall disproportionately on the most disadvantaged. [6]
Pathophysiology
Each genetic test interrogates a different layer of the genome, which is exactly why screens can disagree and why resolution matters. [1] [6]
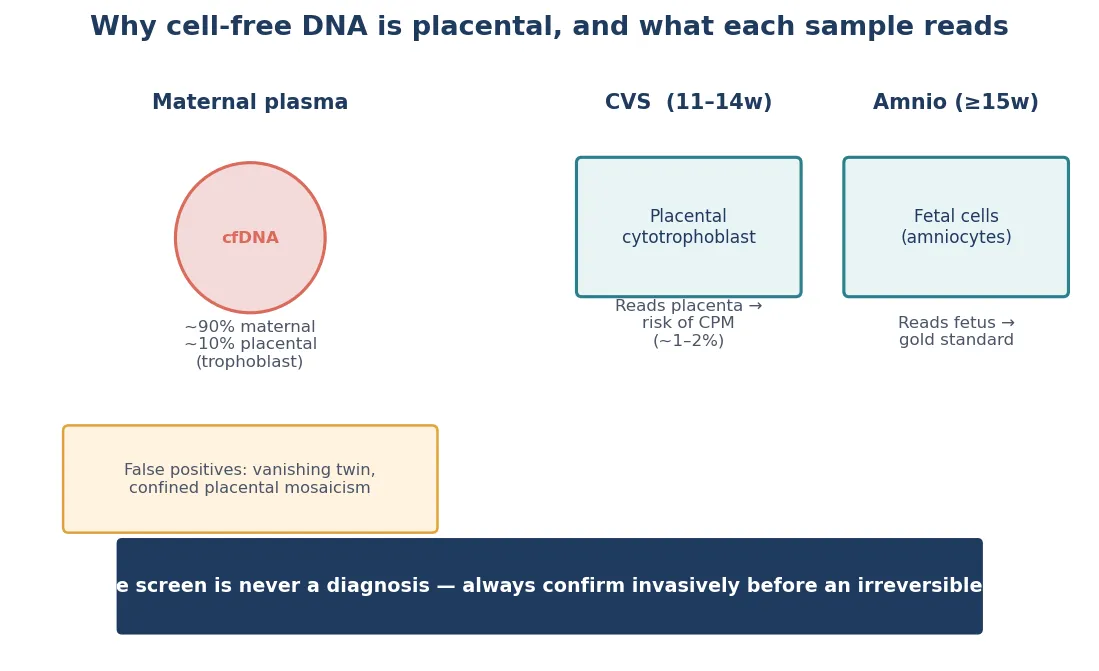
Cell-free DNA works because the placenta sheds DNA fragments into maternal blood. The screening test sequences that mixture and looks for the small fetal fraction. When the fetal fraction is adequate and the fetus carries a common trisomy, the relative excess of that chromosome is detectable. That placental origin matters critically: a confined placental mosaicism can produce a discordant result between cell-free DNA and the true fetal karyotype, which is why cell-free DNA remains a screen. It is also why a no-call is informative — a low fetal fraction is often a biological signal of aneuploidy or placental insufficiency, not just a technical limitation. Cell-based non-invasive prenatal testing, which isolates intact circulating trophoblast cells rather than free DNA fragments, offers the potential for higher-fidelity detection of subchromosomal copy-number variants, though it remains an emerging technology. [3] [10]
Chromosomal microarray detects copy-number variants — submicroscopic deletions and duplications — by comparing patient DNA against reference DNA across millions of probes. Array comparative genomic hybridisation uses fluorescent labelling of patient and reference DNA hybridised to known probes; a single-nucleotide polymorphism array detects genotype at known polymorphic sites and can additionally identify copy-neutral loss of heterozygosity and triploidy. Both technologies resolve copy-number changes far below the karyotype's optical limit, which is the molecular reason microarray increases diagnostic yield in structurally abnormal fetuses. What microarray cannot do is detect balanced rearrangements (which have no net copy-number change), low-level mosaicism below its detection threshold, and single-nucleotide variants. [1] [6]
Exome sequencing interrogates the coding regions — the exons — of approximately 20,000 genes, which harbour approximately 85 per cent of known disease-causing variants. In fetuses with structural anomalies and a normal microarray, exome sequencing adds substantial diagnostic yield because many structural phenotypes are caused by single-nucleotide variants in developmental genes rather than by copy-number changes. Trio exome — sequencing both parents alongside the fetus — helps distinguish de novo variants (often pathogenic) from inherited variants and improves interpretation. Genome sequencing extends coverage to non-coding regions, introns and regulatory elements, and is increasingly used when exome is unrevealing. [11]
Confined placental mosaicism is a pitfall specific to chorionic villus sampling. Because CVS samples trophoblast — the outer layer of the blastocyst that becomes placenta — a chromosomal abnormality present only in the placenta and not in the fetus produces a false-positive CVS result. The rate of confined placental mosaicism is approximately 1 to 2 per cent of CVS samples, and the correct response to a mosaic or unexpected CVS result is confirmation by amniocentesis, which samples fetal cells from amniotic fluid and reflects the true fetal genotype. [4] [6]
Preimplantation genetic testing has its own molecular pitfalls. After in vitro fertilisation, embryo biopsy — typically at the trophectoderm (blastocyst) stage — removes a small number of cells for testing. Whole-genome amplification is required to generate enough DNA from those few cells, which introduces amplification bias. Allele drop-out — the stochastic failure to amplify one allele — can cause a false result in PGT-M if the mutation-bearing allele fails to amplify and the embryo is misclassified as unaffected. Multiple displacement amplification and linkage analysis using informative short tandem repeat markers mitigate but do not eliminate this risk, which is why many programmes recommend prenatal confirmation of PGT-M results by CVS or amniocentesis. [8]
Clinical Presentation
The usual presentation of an abnormal prenatal genetic result is not a sick patient. It is a number on a report, a phone call, or a recall to clinic. The family was expecting reassurance. Your first task is to manage that expectation without minimising or over-calling. [6] [9]
Parents very often hear a positive screen as a confirmed diagnosis. The phrase "your screen came back positive" can be heard as "your baby has Down syndrome." Reframing is a clinical skill: a positive screen means the chance is raised and we need a more accurate test. State what the test does and does not say, then offer the next step. [3] [7]
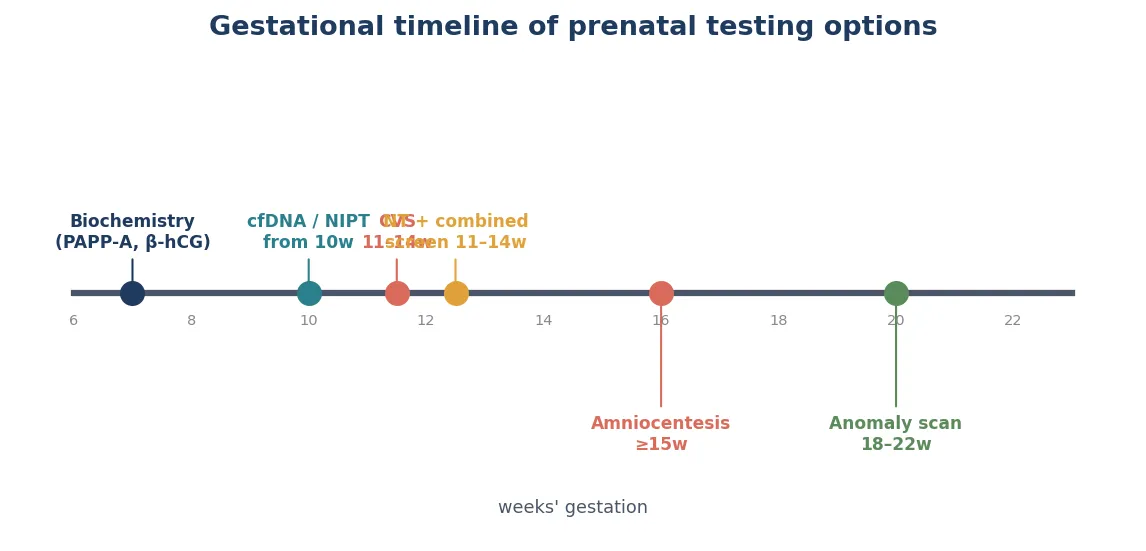
A structural anomaly on the mid-trimester anatomy scan presents as the trigger for diagnostic genetic testing rather than screening alone. When the scan shows a major anomaly — cardiac defect, neural tube defect, diaphragmatic hernia, skeletal dysplasia, or multiple anomalies — the correct cascade is referral to maternal-fetal medicine, detailed sonographic characterisation, and diagnostic testing with chromosomal microarray as first-tier, escalating to exome sequencing if the microarray is normal. The scan is not the end of the diagnostic pathway; it is often the beginning. [1] [9] [11]
A variant of uncertain significance presents as a result that is neither normal nor diagnostic. Families may hear "they found something wrong" or "they don't know if it's a problem." Both framings are hazardous. The correct message is that a variant was found whose clinical meaning is genuinely not yet known by the scientific community, that follow-up testing (parental samples, database review, phenotype correlation) may clarify it, and that the result should not drive irreversible decisions on its own. [1] [6]
A family history of a known Mendelian condition presents at pre-pregnancy or early antenatal counselling. The pathway is targeted: confirm the familial mutation in the affected relative or carrier parent, offer targeted prenatal diagnosis or PGT-M, and involve clinical genetics before rather than after conception where possible. The earlier the mutation is identified, the more reproductive options the family retains. [7] [8]
A confirmed fetal genetic condition presents to the family at multidisciplinary counselling. From that moment the paediatrician's role begins: translate the diagnosis into a postnatal plan, involve neonatology and paediatric surgery and metabolic medicine where relevant, and hold the medical-home role through birth and beyond. For conditions with disease-modifying therapy — for example, some inborn errors of metabolism with available enzyme replacement or dietary management — prenatal diagnosis enables immediate postnatal treatment initiation. [1] [11]
Differential Diagnosis
For a positive aneuploidy screen, four possibilities sit in the differential. A true positive (the fetus does carry the trisomy). A false positive (the screen is positive but the fetus is unaffected — the rate differs by screen type). A screen elevated by a technical or biological confounder such as confined placental mosaicism. And a no-call artefact mistaken for a positive. The discriminating step is diagnostic testing, not a repeat screen when probability is high. [3] [7]
For an increased nuchal translucency with a normal microarray, the differential is broad: cardiac malformation, a genetic syndrome including RASopathies such as Noonan syndrome, structural anomaly, congenital infection and a transient or normal variant. The work-up is detailed fetal echocardiography, full anomaly review, and consideration of exome sequencing or a targeted RASopathy gene panel. A normal microarray does not end the question, because single-gene causes of increased nuchal translucency are invisible to copy-number analysis. [11]
For a pathogenic copy-number variant on prenatal microarray, the critical discrimination is whether the finding is penetrant in this fetus. Some copy-number variants are fully penetrant and define a known syndrome with a predictable phenotype. Others show variable expressivity and incomplete penetrance — the same deletion or duplication may cause severe disease in one individual and nothing in another. Still others are incidental findings unrelated to the sonographic indication. Parental testing helps: if a apparently pathogenic variant is inherited from an unaffected parent, the penetrance question becomes central to counselling. [1] [6]
For a fetus with multiple structural anomalies and a normal microarray, exome or genome sequencing is the next diagnostic step. The diagnostic yield of prenatal exome in this context is substantial — approximately 8 to 10 per cent or higher in well-selected cohorts with normal microarray — and the findings may identify a specific syndrome that drives prognosis, recurrence risk and postnatal management. [11]
For a consanguineous couple with a prior offspring affected by an unidentified autosomal recessive condition, autozygosity mapping combined with exome sequencing is powerful. If the causal gene is identified, PGT-M or targeted prenatal diagnosis becomes available for subsequent pregnancies. If the gene is not identified despite exome, empirical recurrence risk counselling and ongoing ultrasound surveillance apply. [7]
For a cfDNA screen positive for a microdeletion — most commonly 22q11.2 deletion syndrome — the positive predictive value is substantially lower than for common trisomies because the baseline prevalence is lower. The result must be confirmed diagnostically by microarray on a CVS or amniocentesis sample before any management decision. [3] [7]
For a couple with recurrent miscarriages, a parental balanced translocation (reciprocal or Robertsonian) is found in approximately 2 to 5 per cent of couples with three or more losses. Karyotyping both parents identifies the rearrangement, and subsequent reproductive options include PGT-SR or prenatal diagnosis in each pregnancy. [6] [8]
Clinical & Bedside Assessment
Start with the focused family history that changes recurrence risk: maternal and paternal age, parity, prior affected pregnancies or miscarriages, consanguinity, ethnicity, family history of known genetic conditions, developmental delay, congenital anomalies or unexplained deaths, and parental carrier status. These facts move pre-test probability and should be documented before any result is interpreted. [7]
A three-generation pedigree is the cornerstone of reproductive genetics assessment. Construct it systematically, using standard symbols: squares for males, circles for females, a horizontal line for a couple, a vertical line for descent, a diagonal slash for deceased, and a double horizontal line for consanguinity. Shade affected individuals, mark known carriers with a dot, and note twins. A well-drawn pedigree makes recurrence risk calculable and is an exam skill that examiners specifically look for. [7]
Assess the family's real capacity to complete a diagnostic pathway. Who has transport to a tertiary centre? Who has leave from work? Whose phone reliably works for results? Does an interpreter need to be present at the counselling visit, not just a translated letter days later? Who holds decision-making authority within the family structure? A perfect diagnostic booking that no one can attend is not a plan. [6]
At the bedside, the counselling skills are plain language, teach-back, non-directive framing, and explicit separation of screen from diagnosis. State what the test does and does not say. Offer information, options and time. Never force a decision in the same breath as the result. Use teach-back — "Can you tell me in your own words what we just discussed?" — to confirm understanding of a recurrence risk or a variant of uncertain significance, because families under stress retain a fraction of what they hear. [6]
State recurrence risks in the families' terms. For autosomal recessive conditions, the 1-in-4 risk per pregnancy applies when both parents are carriers; emphasise that being a carrier is not the same as being affected. For autosomal dominant conditions, the 1-in-2 risk applies when a parent is affected or carries the pathogenic variant. For X-linked conditions, the risk depends on the sex of the fetus and which parent carries the variant. For mitochondrial conditions, heteroplasmy and the threshold effect complicate prediction. [7] [8]
Investigations
The first-trimester combined screen, done between approximately 11 and 14 weeks, combines nuchal translucency measurement with maternal serum analytes — pregnancy-associated plasma protein A and free beta-human chorionic gonadotropin — alongside maternal age, to estimate risk for the common trisomies. Its detection rate for trisomy 21 is approximately 75 to 85 per cent at a 5 per cent screen-positive rate, which is why it remains the foundational aneuploidy screen in many universal programmes. [7]
Cell-free DNA screening, offered from approximately 10 weeks, analyses placental cell-free DNA in maternal blood. It is highly sensitive for trisomy 21 — detection above 99 per cent with very low false-positive rates in the general-risk population — but its performance for sex-chromosome aneuploidies and microdeletions is lower because the baseline prevalence is lower and the positive predictive value falls. A no-call result is informative rather than neutral. It is a screen, not a diagnostic test, and must not be treated as confirmation. [3] [7]
Chorionic villus sampling, performed from approximately 11 weeks, samples placental trophoblast via a transabdominal or transcervical needle under ultrasound guidance. Its advantage is an earlier result than amniocentesis, which matters for timely reproductive decisions. Its limitations are the risk of confined placental mosaicism — because it samples placental tissue, not fetal cells — and a procedure-related miscarriage risk that is low but real. CVS cannot diagnose neural tube defects or open ventral wall defects because it does not measure amniotic fluid alpha-fetoprotein. [4] [5]
Amniocentesis, performed from approximately 15 to 16 weeks, samples fetal cells desquamated into amniotic fluid via a transabdominal needle under ultrasound guidance. It provides a direct fetal karyotype, microarray, or molecular result and reflects the true fetal genotype without the placental-mosaicism pitfall of CVS. It also allows measurement of amniotic fluid alpha-fetoprotein and acetylcholinesterase for open neural tube defects. Contemporary estimates of procedure-related miscarriage risk are low — contemporary systematic reviews place the additional risk above background at very low levels — and modern, not legacy, figures should be quoted in counselling. [4] [5]
Fetal blood sampling, or cordocentesis, samples fetal blood from the umbilical cord under ultrasound guidance, typically from approximately 18 weeks. Its indications include rapid karyotype (results in 48 to 72 hours), suspected fetal anaemia (parvovirus, Rh alloimmunisation) with measurement of fetal haemoglobin and reticulocytosis, and suspected congenital infection. It carries a higher procedure-related risk than amniocentesis and is reserved for specific questions that cannot be answered from amniotic fluid or placental tissue. [4]
Chromosomal microarray is the recommended first-tier diagnostic test for fetuses with structural anomalies identified on ultrasound. The Wapner trial demonstrated that microarray identified clinically significant copy-number variants in approximately 6 per cent of pregnancies with a normal karyotype and a structural anomaly, which is why it replaced karyotype as the default for this indication. Microarray also detects copy-number variants in pregnancies with no structural anomaly but an increased-risk screen, though the incremental yield is lower. [1] [6]
Prenatal exome sequencing is indicated when a fetus has one or more structural anomalies and the microarray is normal, or when a specific monogenic condition is suspected on phenotypic grounds. The CODE study and related cohorts demonstrate that exome adds diagnostic yield in this context, identifying pathogenic single-nucleotide variants in developmental genes. Trio exome — sequencing both parents and the fetus — is preferred because it improves the interpretation of de novo and inherited variants. Genome sequencing is increasingly used when exome is unrevealing. [11]
Expanded carrier screening uses a multi-gene panel, typically offered to both partners before or early in pregnancy, to identify couples at 1-in-4 risk of an autosomal recessive condition or at risk of an X-linked condition. When both partners are carriers of the same autosomal recessive condition, the reproductive options include PGT-M, prenatal diagnosis each pregnancy, donor gametes, or acceptance of the risk. Counselling before testing should cover the conditions on the panel, residual risk after a negative screen, and the implications of identifying a carrier status. [7]
Preimplantation genetic testing is performed within an in vitro fertilisation cycle. After ovarian stimulation, oocyte retrieval and fertilisation, embryos are cultured to the blastocyst stage (day 5 to 7), and a small number of trophectoderm cells are biopsied. Whole-genome amplification generates enough DNA for testing by linkage analysis (PGT-M), rearrangement-specific testing (PGT-SR), or copy-number analysis (PGT-A). Only embryos classified as unaffected are transferred. PGT-M does not guarantee a live birth — IVF success rates, embryo survival and the stochastic nature of allele drop-out all affect outcomes, and confirmation by prenatal diagnosis is recommended in many programmes. [8]
Non-invasive prenatal diagnosis — as distinct from screening — uses cell-free DNA diagnostically rather than probabilistically for specific questions. Fetal RhD status can be determined from maternal blood in RhD-negative women to guide anti-D prophylaxis. Fetal sex determination from cfDNA is used to guide decisions in X-linked conditions (whether to proceed to invasive testing) and in congenital adrenal hyperplasia (to guide maternal dexamethasone). For selected monogenic conditions, cfDNA can be used diagnostically when the paternal mutation is known and distinguishable from maternal background. These applications are expanding but remain condition-specific. [3] [10]
Management — Resuscitation
Prenatal genetic diagnosis is rarely the first priority in a haemodynamically unstable mother or fetus. Resuscitate first. Once the patient is stable, the genetic question becomes the focus. [4]
The most common "resuscitation" moment in this topic is family crisis. A positive screen, a variant of uncertain significance, or a confirmed pathogenic finding can trigger acute distress. Sit down, use plain language, separate screen from diagnosis, offer written information, involve senior staff and a genetic counsellor, and never force a decision in the same breath as the result. Time is therapeutic. [6]
When prenatal diagnosis identifies a condition with a time-critical postnatal intervention — a ductal-dependent cardiac lesion, a metabolic condition requiring immediate dietary intervention or enzyme replacement, gastroschisis, congenital diaphragmatic hernia — the immediate bundle is multidisciplinary planning and deciding the place and time of delivery so that the right neonatal, surgical and metabolic capability is present. For inborn errors of metabolism with available therapy, prenatal diagnosis can enable diagnostic-ready newborn screening and immediate postnatal treatment, preventing the irreversible damage of a delayed diagnosis. [1] [11]
In rural settings where specialist fetal-medicine, genetics or neonatal capability is not on-site, a significant genetic diagnosis needs destination planning, not optimism. Give a named service, a timeframe and a backup contact before the family leaves. A diagnostic result that goes nowhere is a systems failure. [6]
When a couple receives a pathogenic finding consistent with a non-survivable or profoundly life-limiting condition, the supportive infrastructure — counselling, palliative care planning, perinatal hospice options, and the family's values and beliefs — must be offered without coercion. The paediatrician's role is honest prognosis, symptom-focused planning, and continuity of care regardless of the family's decision. [6]
Management — Definitive & Stepwise
1. Interpret every result against pre-test probability
A result only means something against the prior chance of disease. The same positive cfDNA screen in a 41-year-old and a 19-year-old carries a different positive predictive value. State the pre-test probability before interpreting the result. [3] [7]
2. Low-risk result
Continue routine antenatal care. Counsel residual risk: screening does not exclude every condition, and a normal screen does not exclude single-gene disease. Keep an open door for new concerns. Document explicitly that residual risk was discussed. [7] [9]
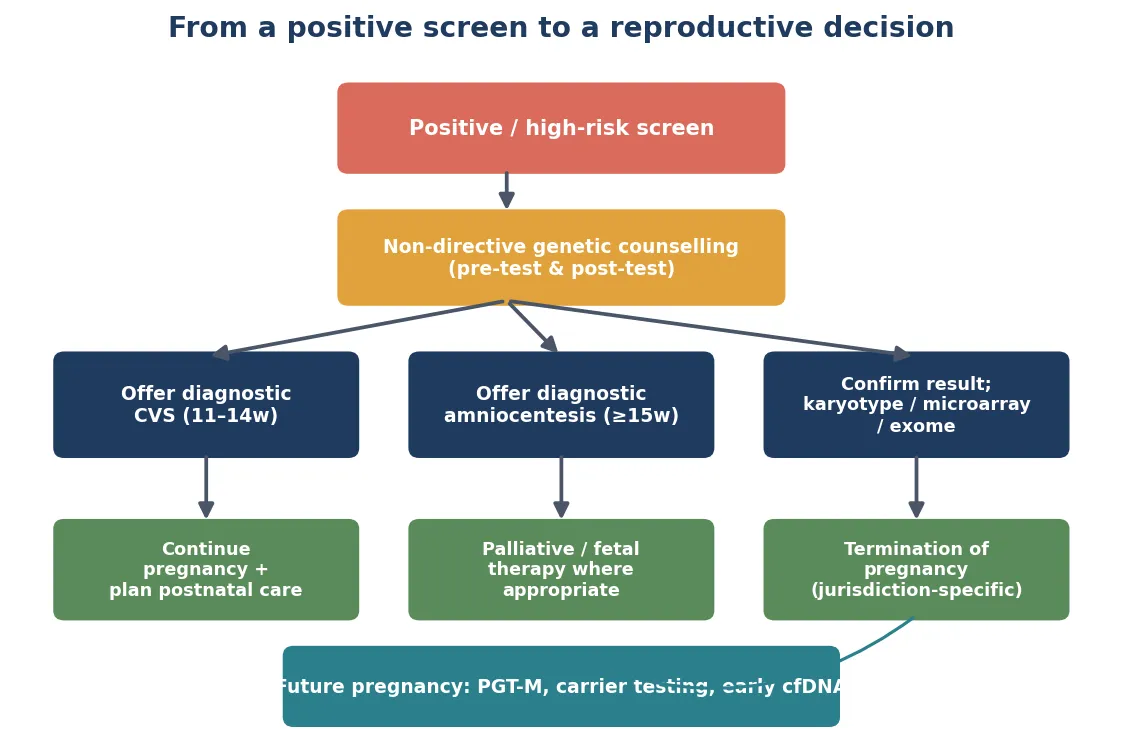
3. Increased-risk result
Offer non-directive counselling and a diagnostic test — CVS or amniocentesis, chosen by gestation and the clinical question. Involve maternal-fetal medicine and clinical genetics. Do not substitute a repeat screen for diagnosis when pre-test probability is genuinely high. For structural anomalies, microarray is first-tier; escalate to exome if microarray is normal. [1] [4] [11]
4. Pathogenic finding
Open the multidisciplinary pathway: maternal-fetal medicine, clinical genetics, neonatology, paediatric surgery, metabolic medicine, and parent support. Correlate genotype with the expected phenotype, counsel prognosis honestly, decide place and time of delivery, prepare the neonatal team, and assign the paediatrician the medical-home role for postnatal surveillance. [1] [11]
5. Variant of uncertain significance
Counsel honestly: this is a real finding whose clinical meaning is not yet established. Offer parental testing (if inherited from an unaffected parent, the penetrance question is central), review against curated databases and phenotype-matching tools, and plan periodic reanalysis as knowledge accumulates. Do not over-call it as pathogenic or dismiss it as benign, and do not let it drive irreversible decisions without further clarification. [1] [6]
6. Known monogenic family risk
Offer PGT-M or natural conception plus targeted prenatal diagnosis each pregnancy. Involve clinical genetics and the IVF team early — before conception where possible, to maximise options. For X-linked conditions, cfDNA fetal sex determination may guide whether invasive testing is needed. For fragile X, counsel on anticipation and full-mutation probability by premutation CGG repeat size. [7] [8]
7. Confirmed fetal condition with reproductive implications
Counsel recurrence risk for future pregnancies. For autosomal recessive conditions (1 in 4 per pregnancy), offer carrier testing of relatives by cascade screening. For autosomal dominant and X-linked conditions, counsel the specific recurrence risk. Discuss PGT-M, prenatal diagnosis, donor gametes and acceptance of risk as the menu of reproductive options, and respect the family's values and decision. [7] [8]
Specific Subtypes & Scenarios
Structural anomaly with normal karyotype. Microarray first. If the microarray is normal and the phenotype suggests a monogenic cause — skeletal dysplasia, cardiac defect with extracardiac anomaly, multiple congenital anomalies — proceed to exome sequencing, ideally as a trio. The diagnostic yield is substantial and may identify a specific syndrome that drives prognosis and postnatal management. [1] [11]
Increased nuchal translucency with normal microarray. A normal copy-number result does not close the question. Detailed fetal echocardiography, full anomaly review, and consideration of a RASopathy gene panel or exome sequencing follow, because Noonan syndrome and related RASopathies are single-gene causes of increased nuchal translucency that are invisible to microarray. [11]
Variant of uncertain significance on microarray or exome. Request parental samples. If the VUS is de novo, it is more likely to be clinically significant. If inherited from an unaffected parent, the penetrance and expressivity question is central. Use curated databases (ClinGen, ClinVar), phenotype-matching tools, and plan periodic reanalysis. Never present a VUS as a diagnosis or as nothing. [1] [6]
Confined placental mosaicism on CVS. Confirm with amniocentesis before acting. A placental-only abnormality does not reflect the fetal genotype, and over-calling it can lead to irreversible decisions based on tissue that is not representative. The rate of confined placental mosaicism on CVS is approximately 1 to 2 per cent. [4] [6]
Consanguineous couple. Enhanced carrier screening, and if a prior child is affected by an unidentified condition, autozygosity mapping combined with exome sequencing. If the causal gene is identified, PGT-M or targeted prenatal diagnosis becomes available. Empirical recurrence risk for first-cousin couples is approximately 2 to 3 per cent above the population baseline for major congenital anomalies and autosomal recessive conditions combined. [7]
Parental balanced translocation. Recurrence risk counselling based on the translocation type and the carrier parent's reproductive history. PGT-SR reduces the risk of aneuploid conceptions and miscarriages. Alternatively, prenatal diagnosis by CVS or amniocentesis in each pregnancy. [6] [8]
Both partners carriers of the same autosomal recessive condition on expanded carrier screening. The 1-in-4 risk per pregnancy applies. Reproductive options include PGT-M, prenatal diagnosis each pregnancy, donor gametes, and acceptance of risk. Genetic counselling before pregnancy maximises options. [7] [8]
Fragile X premutation carrier mother. The risk of expansion to a full mutation in offspring depends on the premutation CGG repeat size — larger premutations are more likely to expand to full mutations that cause fragile X syndrome. PGT-M and prenatal diagnosis are available. Counsel on the spectrum from premutation (risk of fragile X-associated tremor/ataxia syndrome and primary ovarian insufficiency) to full mutation (intellectual disability, autism features). [7] [8]
Mitochondrial conditions. Heteroplasmy, the threshold effect, and mitotic segregation make recurrence risk prediction complex. PGT for mitochondrial disease is possible in selected cases but has limitations. Nuclear transfer techniques (so-called three-parent IVF or mitochondrial donation) are available for a narrow set of conditions in some jurisdictions and represent a frontier of reproductive genetics. [8]
Complications & Pitfalls
False-positive screens cause anxiety, extra visits and, in the worst case, an unnecessary invasive procedure with its own risk. Accurate non-directive counselling and rapid access to diagnostic testing minimise this harm. The psychological burden of a false-positive screen is substantial and should not be minimised. [3] [4]
False-negative and missed diagnoses create the opposite harm: an unanticipated finding at birth, lost preparation time, and a neonatal team caught unready. For monogenic conditions not interrogated by standard aneuploidy screening, the residual risk after a normal screen is real and should be counselled. [1] [11]
Over-interpretation of a variant of uncertain significance is the modern equivalent of the false positive. As exome and genome sequencing become routine, the volume of VUS findings rises, and the counselling burden grows. The correct response is honest uncertainty, parental testing, database review and reanalysis — not reflexive action. [1] [6]
Classic pitfalls recur across exams and practice: treating a screen as a diagnosis, relabelling a no-call as low-risk, over-calling a VUS, quoting legacy procedure-related risks, reporting confined placental mosaicism from CVS without confirmation, discharging an inconclusive result with no owner, forgetting residual risk after a normal diagnostic result, and failing to confirm PGT-M results prenatally. [1] [4] [8]
Lost-to-follow-up is the quiet complication that destroys programme benefit. A pathogenic or VUS result with no booked next appointment is a systems failure and should be chased like any abnormal critical result. Poor documentation and handover across sites and clinicians compounds the problem. [6]
Inequitable access to PGT and advanced genetic testing is a structural complication. Cost, geography and regulatory variation mean that the families who would benefit most from PGT-M or exome sequencing are often the least able to access it. The paediatrician's advocacy role includes awareness of local funding pathways and equity of referral. [8]
Prognosis & Disposition
Prognosis after an abnormal prenatal genetic result is governed by three things: the pre-test probability, the positive predictive value of the result in that individual, and whether diagnostic testing confirms a real condition. A screen raises concern; only confirmation defines prognosis. [3] [7]
A pathogenic finding for a well-characterised syndrome has a prognosis defined by the natural history of that condition — and the paediatrician's job is to translate that natural history into a postnatal plan the family can understand and act on. For conditions with disease-modifying therapy, prenatal diagnosis can change the prognosis by enabling immediate postnatal treatment. [1] [11]
A variant of uncertain significance has a prognosis that is genuinely unknown. Honesty is the correct disposition: state what is known, what is unknown, what follow-up might clarify, and avoid presenting uncertainty as either reassurance or alarm. [1] [6]
A normal microarray or exome does not exclude all genetic disease. Residual risk persists for single-gene conditions not on the tested panel, regulatory variants not captured by exome, mosaicism below detection thresholds, and structural disease. Residual-risk counselling after a normal diagnostic result is mandatory. [6] [11]
Disposition is "result owned, loop closed." A low-risk result goes home with routine care and an open door. An increased-risk result goes home only with a booked diagnostic pathway and a named owner. A pathogenic finding goes home with a documented multidisciplinary plan and a neonatal alert. A VUS goes home with honest counselling, parental testing arranged and a reanalysis plan. A declined result goes home with documented residual risk and an open door. [1] [6]
PGT-M does not guarantee a live birth. IVF success rates, embryo survival, embryo mosaicism and the stochastic nature of allele drop-out all affect outcomes. Couples pursuing PGT-M need honest counselling about cumulative success rates and the emotional and financial cost of repeated cycles. [8]
Special Populations
Consanguineous couples carry an enhanced autosomal recessive risk. First-cousin unions add approximately 2 to 3 per cent above the population baseline for major congenital anomalies and serious recessive conditions combined. Enhanced carrier screening, autozygosity mapping if a prior child is affected, and culturally safe counselling are essential. Stigmatising consanguinity is counterproductive; the goal is risk assessment and informed choice. [7]
Indigenous, Māori, Pacific, migrant, refugee and asylum-seeking families need interpreter-supported counselling at the moment of the result, not a translated letter days later, and care that respects family decision structures. Access barriers — transport, leave, language, trust — reduce screening and diagnostic completion in these populations and must be designed around, not wished away. [6]
Couples from populations with high carrier frequency for specific founder conditions benefit from targeted carrier screening: Tay-Sachs and other conditions in Ashkenazi Jewish populations, sickle cell disease in West African populations, beta-thalassaemia in Mediterranean and South Asian populations, cystic fibrosis in Northern European populations. Expanded pan-ethnic screening captures most of these without relying on self-reported ethnicity. [7]
Advanced maternal age raises aneuploidy pre-test probability and is the classic risk factor for trisomy 21. Advanced paternal age raises the risk of de novo dominant mutations — achondroplasia, Apert syndrome and other FGFR-related conditions — by a small but measurable increment. Both shift the counselling but neither defines a high-risk pregnancy on its own. [3] [7]
Families connected to Deaf or disability communities may frame genetic outcomes differently from clinicians. Good care is accurate information, timely diagnosis, and respect for family values — never ableism dressed up as counselling. A positive diagnosis of a condition associated with disability is not uniformly a negative outcome, and non-directive counselling means presenting the full range of perspectives. [6]
Rural and remote families need diagnostic pathways matched to real travel times and local capability. Socioeconomic disadvantage reduces access to advanced genetic testing and PGT. Couples with infertility requiring IVF face cumulative cost, and PGT adds to that burden with regulatory and funding variation across jurisdictions. [8]
Evidence, Guidelines & Regional Differences
The Wapner trial (NEJM 2012) established chromosomal microarray as superior to karyotype for prenatal diagnosis of structurally abnormal fetuses, identifying clinically significant copy-number variants in approximately 6 per cent of cases with normal karyotype. This single trial shifted the standard of care. The Reddy NICHD stillbirth study, published in the same issue, extended microarray utility to stillbirth evaluation. [1] [2]
The CARE trial (Norton, NEJM 2015) demonstrated that cell-free DNA screening had high sensitivity and specificity for trisomy 21 in a general-risk population, which is the evidence base for offering cfDNA beyond high-risk cohorts. Its limitations — lower performance for sex-chromosome aneuploidies and microdeletions, the meaning of a no-call — are equally well described. [3]
Contemporary systematic reviews (Salomon 2019, Beta 2019) placed the procedure-related miscarriage risk of CVS and amniocentesis at low levels and argued against quoting legacy, higher estimates that deter families from diagnostic testing they need. These reviews are the evidence base for counselling modern, low procedure-related risk. [4] [5]
The ESHRE PGT Consortium data collections define international PGT activity, outcomes and trends, and are the primary source for PGT-M, PGT-SR and PGT-A volume and success data. [8]
The CODE study (Lord et al, 2021) demonstrated the diagnostic yield of prenatal exome sequencing in congenital heart disease and has contributed to the evidence base for exome in structurally abnormal fetuses with normal microarray. [11]
Australia and Aotearoa New Zealand offer universal aneuploidy and anomaly screening under national pregnancy-care guidance, with the Human Genetics Society of Australasia providing carrier screening, prenatal diagnosis and PGT guidance. Access to publicly funded cfDNA and expanded carrier screening varies by jurisdiction, and PGT funding is limited. Quote the local programme panels and windows. [7]
United Kingdom runs national antenatal screening programmes (NHS) with defined offer, referral and diagnostic pathways, and the Royal College of Obstetricians and Gynaecologists provides green-top guidance on prenatal diagnosis. The Society for Histocompatibility and Genomics in the UK and regional genetics centres manage molecular testing. [7]
United States follows ACOG and ACMG guidance; cfDNA is offered to all pregnant patients regardless of risk, and microarray is first-tier for structurally abnormal fetuses. PGT is widely available through commercial providers with variable insurance coverage. [1] [7]
Canada uses provincial programmes; geography and access often dominate diagnostic planning, and the Canadian College of Medical Geneticists provides testing standards. [7]
State only differences you have verified against current official programme pages. Do not invent a fixed screening window, a universal carrier screening panel, or a PGT success rate that is not sourced. [4]
Exam Pearls
- Screening estimates risk; diagnostic testing confirms. Never blur the two. [3]
- Microarray is first-tier for structurally abnormal fetuses; exome if microarray is normal. [1] [11]
- CVS samples placenta; amniocentesis samples fetal cells. Confined placental mosaicism is a CVS pitfall — confirm with amniocentesis. [4] [6]
- A variant of uncertain significance is a real finding whose meaning is genuinely unknown — never over-call. [1]
- PGT-M does not replace prenatal diagnosis — recommend confirmation in many programmes. [8]
- Normal microarray or exome does not exclude all genetic disease — counsel residual risk. [6] [11]
CONFIRM package
References
- [1]Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. The New England journal of medicine, 2012.PMID 23215555
- [2]Reddy UM, Page GP, Saade GR, et al. Karyotype versus microarray testing for genetic abnormalities after stillbirth. The New England journal of medicine, 2012.PMID 23215556
- [3]Norton ME, Jacobsson B, Swamy GK, et al. Cell-free DNA analysis for noninvasive examination of trisomy. The New England journal of medicine, 2015.PMID 25830321
- [4]Salomon LJ, Sotiriadis A, Wulff CB, et al. Risk of miscarriage following amniocentesis or chorionic villus sampling: systematic review of literature and updated meta-analysis. Ultrasound in obstetrics & gynecology, 2019.PMID 31124209
- [5]Beta J, Zhang W, Geris S, et al. Procedure-related risk of miscarriage following chorionic villus sampling and amniocentesis. Ultrasound in obstetrics & gynecology, 2019.PMID 30977213
- [6]Stosic M, Levy B, Wapner R. The use of chromosomal microarray analysis in prenatal diagnosis. Obstetrics and gynecology clinics of North America, 2018.PMID 29428286
- [7]American College of Obstetricians and Gynecologists. Screening for Fetal Chromosomal Abnormalities: ACOG Practice Bulletin Summary, Number 226. Obstetrics and gynecology, 2020.PMID 32976375
- [8]Spinella F, Bronet F, Carvalho F, et al. ESHRE PGT Consortium data collection XXI: PGT analyses in 2018. Human reproduction open, 2023.PMID 37091225
- [9]Salomon LJ, Alfirevic Z, Berghella V, et al. ISUOG Practice Guidelines (updated): performance of the routine mid-trimester fetal ultrasound scan. Ultrasound in obstetrics & gynecology, 2022.PMID 35592929
- [10]Vossaert L, Wang Q, Salman R, et al. Reliable detection of subchromosomal deletions and duplications using cell-based noninvasive prenatal testing. Prenatal diagnosis, 2018.PMID 30357877
- [11]Lord J, McMullan DJ, Eberhardt RY, et al. Congenital heart disease and the Diagnostic yield with Exome sequencing (CODE) study: prospective cohort study and systematic review. Ultrasound in obstetrics & gynecology, 2021.PMID 32388881