Paeds · investigations-procedures-and-technology
Genetic and metabolic test selection
Also known as Choosing a genetic test · Microarray versus exome · Newborn bloodspot screening · Tandem mass spectrometry screening · Metabolic workup in a child · Chromosomal microarray first-tier · Rapid genomic sequencing in the NICU
Fellowship guide to choosing the right genetic and metabolic test in a child, owned from the requester's side of the form rather than the laboratory's side of the bench. Covers the resolution ladder from karyotype through chromosomal microarray to whole-exome and whole-genome sequencing, the diagnostic yields of each platform in developmental delay, congenital anomalies, autism and the critically ill infant, the design and interpretation of newborn bloodspot screening by tandem mass spectrometry, the metabolic test panel — plasma amino acids, urine organic acids, acylcarnitines, lactate, transferrin isoforms and very-long-chain fatty acids — and how to take consent, request a trio, interpret a variant of uncertain significance, manage an incidental secondary finding, and apply the ACMG, HGSA, RCPCH and regional newborn screening programmes to real children.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the four-year-old in your general paediatric clinic with unexplained global developmental delay and a normal microarray, the neonate in the NICU with hypotonia, seizures and an uninformative septic screen, and the baby brought back at two weeks because a spot of blood from the heel-prick at 72 hours has returned an elevated octanoylcarnitine. The shared question is which tube to fill, in which child, in which window, and what to do with the answer. That question is what this page owns — not the platforms in depth (owned by the chromosomal microarray, exome and genome sequencing page) and not the diseases themselves, but the act of choosing and interpreting the test in a real child. [1] [10]
Genetic test selection is the request-side skill of matching a clinical question to a platform — karyotype for suspected aneuploidy or balanced rearrangements; chromosomal microarray for copy-number variants in developmental delay, intellectual disability, autism with dysmorphism, and multiple congenital anomalies; whole-exome and whole-genome sequencing for the single-nucleotide variants, small indels and (for genome) the non-coding variants that cause most Mendelian disease. Metabolic test selection is the parallel skill of matching a metabolic suspicion to the right analyte in the right sample in the right window — plasma amino acids, urine organic acids, the acylcarnitine profile, lactate and pyruvate, transferrin isoforms for congenital disorders of glycosylation, and very-long-chain fatty acids for peroxisomal disease. Newborn bloodspot screening is a third and distinct entity: a population screen, not a diagnostic test, run from a heel-prick onto a dried blood spot, that flags the infant who needs a confirmatory diagnostic test. [1] [12] [14]
The discipline that ties all three together is the Wilson and Jungner principles of screening — the condition must be an important health problem, there must be an accepted treatment, facilities for diagnosis and treatment must be available, there must be a recognisable latent or early symptomatic stage, and there must be a suitable test — applied honestly, so that a screen does not become a diagnostic label and a diagnostic test does not become a screen. [12]
Classification
Sort genetic and metabolic tests by what they detect, their resolution, the sample they need, and the population they serve — because those four axes decide which test answers the clinical question at the lowest cost in time, money and family harm. [1] [2]
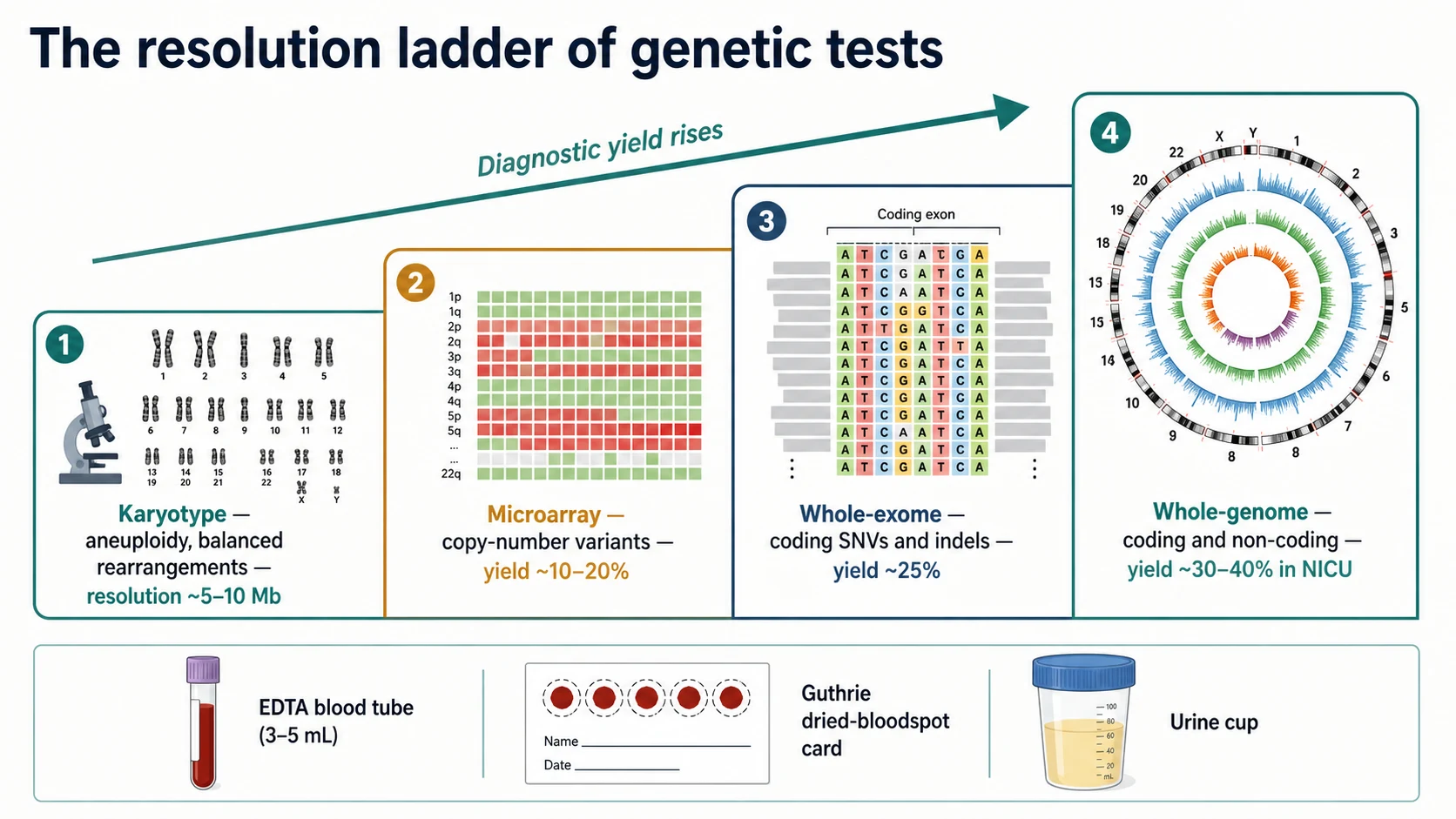
A karyotype cultures the child's lymphocytes, arrests them in metaphase, stains the chromosomes, and images them down a light microscope at a resolution of about 5 to 10 megabases. It detects aneuploidy (trisomy 21, 18, 13; Turner and Klinefelter syndromes), and large balanced rearrangements such as reciprocal translocations and inversions that a microarray cannot see. Its modern role is narrow: a suspected aneuploidy or a known familial balanced rearrangement, and the workup of recurrent miscarriage. It is not the first-tier test for unexplained developmental delay. [1]
A chromosomal microarray (comparative genomic hybridisation array, or a single-nucleotide-polymorphism array) compares the child's DNA to a reference across millions of points along the genome and flags copy-number variants — submicroscopic deletions and duplications — at a resolution down to a few hundred kilobases, far finer than a karyotype. The 2010 consensus statement of Miller and colleagues established microarray as the first-tier clinical diagnostic test for the child with unexplained developmental delay, intellectual disability, autism with dysmorphism, or multiple congenital anomalies, replacing the karyotype in that role. Its yield in this population is on the order of 10 to 20 percent. It does not detect balanced rearrangements, low-level mosaicism, single-nucleotide variants, or repeat expansions, so a normal microarray does not exclude a genetic diagnosis — it tells you to move up the ladder. [1]
Whole-exome sequencing captures and sequences the coding exons of the roughly 20,000 genes — about one percent of the genome — where most disease-causing single-nucleotide variants and small insertions or deletions live. Yang and colleagues' 2013 series established clinical exome sequencing as a diagnostic tool with an overall yield of around 25 percent, and it has become the second-tier test after a normal microarray in the child with a suspected monogenic disorder. Whole-genome sequencing sequences coding and non-coding regions alike, capturing the regulatory and structural variants that an exome misses; it is the platform of choice for the acutely ill infant in the NICU and for the exome-negative diagnostic odyssey. [2] [3]
Microarray versus exome versus genome at a glance
Chromosomal microarray
- Detects copy-number variants (deletions, duplications) — not single-nucleotide variants
- First-tier test for unexplained DD, ID, autism with dysmorphism, multiple congenital anomalies
- Diagnostic yield about 10 to 20 percent in this population
- Does not detect balanced rearrangements, low-level mosaicism, repeat expansions
- EDTA blood, 3 to 5 mL; turnaround weeks; do not repeat once normal
Whole-exome sequencing
- Detects coding single-nucleotide variants and small indels across ~20,000 genes
- Second-tier after a normal microarray in a suspected monogenic disorder
- Diagnostic yield about 25 percent ambulant, 30 to 40 percent in the critically ill infant
- Trio (child plus both parents) roughly doubles yield over proband-only
- Can return a VUS, an incidental actionable finding, or a non-paternity result — consent first
Whole-genome sequencing
- Detects coding and non-coding variants, structural and repeat variants exome misses
- Platform of choice for the acutely unwell neonate and the exome-negative odyssey
- Rapid turnaround (days) is available for the critically ill infant
- Higher data burden, higher cost, deeper reanalysis potential over time
- Same consent considerations as exome; secondary findings list applies
Metabolic tests are classified by the analyte they measure and the disorder family they pick up. Plasma amino acids flag the aminoacidopathies (phenylketonuria, maple syrup urine disease, the urea-cycle disorders). Urine organic acids flag the organic acidaemias (methylmalonic, propionic, isovaleric). The plasma acylcarnitine profile flags the fatty-acid oxidation disorders (medium-chain acyl-CoA dehydrogenase deficiency) and several organic acidaemias. Lactate and pyruvate flag mitochondrial and pyruvate-metabolism disease. Transferrin isoforms (by isoelectric focusing or mass spectrometry) flag the congenital disorders of glycosylation. Very-long-chain fatty acids flag peroxisomal disorders. Newborn bloodspot screening, by contrast, is not one of these panels — it is a dried-blood-spot multianalyte screen run on a tandem mass spectrometer that flags the child who then needs the diagnostic panel. [12] [14]
The numbers that anchor your viva
Epidemiology & Risk Factors
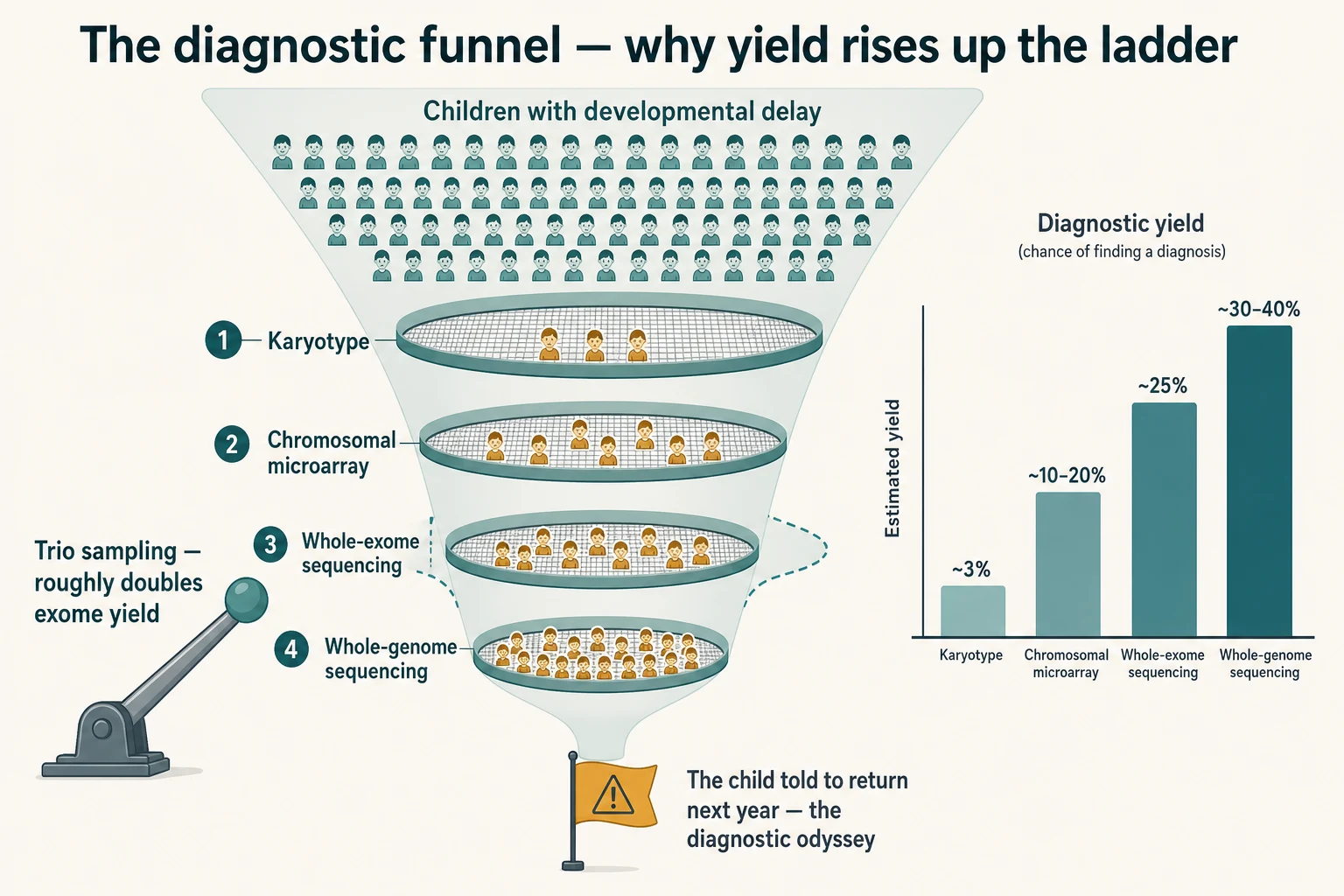
Genetic disease is common in paediatrics, and the question is not whether to test but how to test well. About 2 to 5 percent of newborns carry a major congenital anomaly, a single-gene disorder, or a chromosomal abnormality, and genetic disease accounts for a disproportionate share of paediatric intensive-care admissions and paediatric deaths. The child who arrives at the request form is the child who carries a real burden of genetic disease, and the test selection is the lever that moves that burden into a diagnosis. [1] [8]
The child at greatest likelihood of a positive genetic test is the one whose clinical picture concentrates the prior probability. Multiple congenital anomalies with developmental delay, global developmental delay with regression or dysmorphism, intellectual disability of unexplained cause, autism spectrum disorder with intellectual disability or dysmorphism, a family history of consanguinity or of a compatible recessive condition, and a sibling with a similar phenotype are the populations in whom the prior probability is high enough that the test will often answer. The Tammimies autism series found that the combined yield of microarray and exome was around 16 percent in children with autism spectrum disorder, with the yield concentrated in those with dysmorphism or intellectual disability; the well, non-dysmorphic child with isolated autism has a lower yield, and that prior shapes the request. [1] [5]
The child at greatest risk of being missed is the one at the wrong end of the diagnostic odyssey. Sawyer and colleagues found that the average child referred for whole-exome sequencing near the end of the diagnostic odyssey had spent years, seen multiple specialists, and accumulated a substantial number of uninformative tests before reaching the test that answered. The population-level problem is one of late and under-sequenced referral — the child who has a normal microarray and is told to come back in a year, rather than being escalated to exome. [6] [9]
Pathophysiology
To explain why the resolution ladder rises in yield, follow the variant through the platforms. A copy-number variant — a deletion or duplication of hundreds to millions of base pairs — is invisible to a karyotype when it is smaller than the microscope's resolution, but a microarray sees it because the array probes millions of points and reports a relative gain or loss against the reference. A single-nucleotide variant — a change in one base pair in one exon of one gene — is invisible to both karyotype and microarray, because neither reads the sequence letter by letter; an exome or genome reads the sequence and reports the variant against a reference. The platforms are not interchangeable; they detect different classes of variant, and that is why a normal microarray does not exclude a genetic diagnosis. [1] [2]
The other half of the pathophysiology is the de novo variant. The single most powerful signal in exome interpretation is the variant that is present in the affected child and absent from both unaffected parents — a variant that arose in the germline of one parent or in early embryogenesis. A trio sample (child and both parents) lets the bioinformatician filter the 20,000 or so coding variants found in a typical child down to the small handful that are de novo and plausibly disease-causing, which is why trio sequencing roughly doubles the diagnostic yield of proband-only exome. A proband-only sample, in a child with no informative family structure, leaves a much larger variant list that the laboratory must prioritise by phenotype. [2] [4]
The metabolic platforms have their own physiology. Tandem mass spectrometry, the engine of expanded newborn bloodspot screening, ionises each analyte, separates it by mass-to-charge ratio in a first mass analyser, fragments it, and separates the fragments in a second mass analyser; the result is a quantitative fingerprint of dozens of acylcarnitines and amino acids from a single punch of a dried blood spot. The reason the screening test is taken at 48 to 72 hours of life is metabolic: the analytes the screen measures (phenylalanine, leucine and isoleucine, the acylcarnitines) only become informative once the infant has been feeding and metabolising for a day or two, and they become misleading if collected too early or after a transfusion. [12] [14]
[12] [14]Clinical Presentation
The child who needs a genetic or metabolic test comes to the request form through one of a small number of presentations, and the presentation shapes the test. [1] [12]
The dysmorphic neonate with multiple congenital anomalies asks: is there a unifying chromosomal or single-gene diagnosis. The child with global developmental delay or intellectual disability of unexplained cause asks: is there a copy-number variant or a monogenic disorder. The child with regression asks: is there a neurodegenerative or neurometabolic cause. The child with an abnormal newborn bloodspot screen asks: is this real, and what is the diagnosis. The critically ill infant in the NICU with an uninformative septic and metabolic screen asks: is there a monogenic disorder driving the decompensation. Each is a different clinical question, and each routes to a different test. [1] [8]
The red-flag presentations that raise the priority of testing — and shift the choice toward rapid sequencing rather than sequential single-gene tests — are a critically ill infant with a suspected monogenic disorder, a child with regression, a family history of consanguinity with a compatible phenotype, a previously affected sibling, and the dysmorphic neonate with major anomalies who is unstable. These are the children in whom the yield is highest and the management consequence is greatest, and the request should be escalated, not deferred. [7] [8]
The atypical and difficult presentations deserve attention. The child of consanguineous parents carries a higher prior probability of an autosomal recessive condition, and the request should consider an exome or genome rather than sequential targeted tests. The adopted or migrant child without an informative family history loses the trio advantage and the segregation analysis, so the phenotype must drive the request. The neonate with a suspected inborn error of metabolism in metabolic decompensation is the child for whom the window is everything — the metabolic panel sent after recovery is often normal, and the sample taken during the decompensation is the diagnostic one. [4] [12]
Differential Diagnosis
The differential for the request is best framed as the diagnostic question the test must answer — for each presentation, what is the test looking for, and what platform finds it. [1] [2]
In the child with global developmental delay and a normal microarray, the differential is a monogenic neurodevelopmental disorder, a non-coding regulatory variant, a repeat expansion, or a metabolic cause; the test is whole-exome or whole-genome sequencing, sent as a trio where both parents are available. Lee and colleagues and Yang and colleagues established that exome finds the diagnosis in around a quarter of these children after a normal microarray. [2] [3]
In the child with regression, the differential is a neurometabolic cause (a lysosomal storage disorder, a leukodystrophy, a mitochondrial disorder), a neurodegenerative cause, or an epileptic encephalopathy; the test sequence is metabolic first (urine organic acids, plasma amino acids, acylcarnitines, lactate, very-long-chain fatty acids, transferrin isoforms), then brain MRI, then exome or genome for the child in whom the metabolic and imaging workup is unrevealing. A normal metabolic panel during a well interval does not exclude a metabolic cause. [12]
In the critically ill infant with an uninformative septic and metabolic screen, the differential is a severe monogenic disorder driving the decompensation; the test is rapid whole-genome sequencing as a trio, which the NSIGHT1 randomised controlled trial of Petrikin and colleagues showed yields a diagnosis in around 30 to 40 percent of these infants within days, against around 3 percent with standard testing. The management consequence is real — withdrawing intensive care, escalating a specific therapy, redirecting to palliation, or avoiding a harmful procedure. [7] [8]
In the abnormal newborn bloodspot screen, the differential is the small set of disorders the screen measures — and the action is the confirmatory diagnostic test, not the assumption of disease. An elevated phenylalanine triggers quantitative plasma amino acids for phenylketonuria; an elevated octanoylcarnitine triggers plasma acylcarnitines and a metabolic specialist referral for medium-chain acyl-CoA dehydrogenase deficiency; an immunoreactive trypsinogen elevation triggers a sweat chloride or CFTR genetic test for cystic fibrosis; an elevated 17-hydroxyprogesterone triggers a confirmatory serum test for congenital adrenal hyperplasia; an abnormal thyroxine or thyroid-stimulating hormone triggers confirmatory thyroid function tests for congenital hypothyroidism. The ACMG ACT sheets give the algorithm for each. [12] [13]
Clinical & Bedside Assessment
Before any genetic or metabolic test, run a structured bedside assessment that turns the child's presentation into the right test and the right sample. [1] [12]
Begin with a three-generation pedigree, drawn at the bedside with the symbols right. Record consanguinity explicitly (a question parents may not volunteer), parental ages, ethnic origins, recurrent miscarriages, stillbirths, neonatal deaths, and affected relatives. Take a focused history of the pregnancy, the perinatal period, the feeding and developmental milestones, and any regression — the regression is the single most important clue that lifts the priority of a metabolic and neurodegenerative workup. Examine the child for dysmorphism, with measurements where relevant (occipitofrontal circumference, height, span, upper-to-lower segment ratio), and document the neurocutaneous stigmata, the organomegaly (hepatosplenomegaly flags storage disease), the tone and the deep tendon reflexes, and the developmental profile against age-appropriate norms. [1]
The assessment of the metabolic window is the step most often missed. A child who is decompensating with acidosis, hyperammonaemia, hypoglycaemia or encephalopathy is the child for whom the acute sample set must be drawn before treatment blunts the abnormality: venous blood gas, glucose, ammonia (on ice, to the laboratory immediately), lactate, beta-hydroxybutyrate, plasma amino acids, urine organic acids (any urine is better than no urine), a free-carnitine and acylcarnitine profile, and a saved aliquot of plasma and urine at minus 80 for later testing. Treating first and sampling later closes the diagnostic door, because most metabolites normalise with dextrose, protein restriction and detoxification. [12]
The assessment of stability for the request is the step that decides the platform. The well child in the outpatient clinic gets an elective exome or microarray with a turnaround of weeks. The critically ill infant in the NICU gets a rapid whole-genome sequencing with a turnaround of days, because the diagnostic answer changes the management within the window it can act on — and Soden and colleagues showed, in their acuity-stratified series, that the yield and the management impact of sequencing are highest in the sickest children. [4] [8]
Investigations
The investigations section of this topic is the test-selection ladder itself, reproduced as the examinable decision rules — because the rules are the content the fellowship tests. The two pillars are the microarray-first consensus for the ambulant child with developmental delay, and the rapid whole-genome sequencing paradigm for the critically ill infant, with newborn bloodspot screening as the population-level screen that stands alongside. [1] [8] [12]
The genetic test ladder
The genetic test selection ladder
Suspected aneuploidy or a known familial balanced rearrangement — karyotype (rapid, detects whole-chromosome and large balanced changes; not the test for developmental delay)
Unexplained global developmental delay, intellectual disability, autism with dysmorphism, or multiple congenital anomalies — chromosomal microarray as first-tier (yield ~10–20%)
Microarray normal and a monogenic disorder still suspected — whole-exome sequencing as a trio where possible (yield ~25%, roughly doubled by trio sampling)
Critically ill infant with a suspected monogenic disorder, or exome-negative diagnostic odyssey — rapid whole-genome sequencing as a trio (yield ~30–40% in the NICU)
All genetic tests — document the phenotype, take consent, request the trio, plan the result communication and the cascade testing
The Miller 2010 consensus statement is the foundational reference for the microarray-first position. It established that, for the child with unexplained developmental delay or intellectual disability, an autism spectrum disorder with dysmorphism, or multiple congenital anomalies, chromosomal microarray is the first-tier test because it detects the submicroscopic copy-number variants that a karyotype misses, with a diagnostic yield of around 10 to 20 percent against a karyotype yield of around 3 percent in the same population. A karyotype is reserved for the suspected aneuploidy and the known familial balanced rearrangement. [1]
The Tan and colleagues ambulant cohort and the Yang exome series established the second tier. For the child with a normal microarray and a suspected monogenic disorder, whole-exome sequencing finds the diagnosis in around 25 percent of cases, with the yield rising in the child with a clearer monogenic phenotype, with intellectual disability, and with a trio sample. Tan and colleagues also found that the cost per diagnosis fell when exome was used earlier rather than later in the workup, which is the economic argument against the diagnostic odyssey. [2] [9]
The NSIGHT1 randomised controlled trial of Petrikin and colleagues, and the ICU exome series of Meng and colleagues, established the rapid paradigm for the critically ill infant. Rapid whole-genome sequencing in the NICU yields a diagnosis in around 30 to 40 percent of infants with a suspected monogenic disorder, against around 3 percent with standard testing, and the diagnosis changes management in a meaningful proportion — withdrawing intensive care, escalating a specific therapy such as a cofactor, or redirecting to palliation. The acuity of the child drives the speed of the platform. [7] [8]
| Clinical question | First-line test | Why, and the caveat |
|---|---|---|
| Suspected aneuploidy (Down, Turner, Klinefelter) or known familial translocation | Karyotype (or rapid QF-PCR for urgent aneuploidy) | Sees whole chromosomes and balanced rearrangements; not the test for developmental delay (Cite id=1). |
| Unexplained global developmental delay, intellectual disability | Chromosomal microarray first-tier | Miller 2010 consensus; yield ~10–20%; do not repeat once normal — escalate to exome (Cite id=1,2). |
| Autism with dysmorphism or intellectual disability | Chromosomal microarray, then exome | Tammimies 2015 combined yield ~16%; isolated non-dysmorphic autism has lower yield (Cite id=5). |
| Multiple congenital anomalies | Chromosomal microarray, then exome or genome | Yield rises with the number of anomalies; rapid genome in the dysmorphic unstable neonate (Cite id=1,8). |
| Normal microarray, suspected monogenic disorder | Whole-exome sequencing as a trio | Yang 2013 yield ~25%; trio doubles yield over proband-only; consent for VUS and secondary findings (Cite id=2). |
| Critically ill infant, suspected monogenic disorder | Rapid whole-genome sequencing as a trio | NSIGHT1 RCT yield ~30–40% vs ~3% standard; changes management in days (Cite id=7,8). |
| Exome-negative diagnostic odyssey | Whole-genome sequencing or exome reanalysis | Genome captures non-coding and structural variants exome misses; reanalyse every 1–2 years (Cite id=6). |
| Suspected repeat-expansion disorder (Fragile X, myotonic) | Targeted repeat-primed PCR or Southern blot | Exome and genome may miss pathogenic expansions — request the specific test (Cite id=1). |
Newborn bloodspot screening
Newborn bloodspot screening is the population-level screen that runs in parallel to the diagnostic ladder, and the requesting paediatrician owns its execution and its interpretation. The sample is a heel-prick onto a Guthrie card, taken at 48 to 72 hours of life, with the blood applied to one side of the card and allowed to soak through, never squeezed or layered. The platform is tandem mass spectrometry, which measures dozens of acylcarnitines and amino acids from a single punch, alongside immunoassays for thyroxine or thyroid-stimulating hormone, immunoreactive trypsinogen, and the haemoglobinopathies, and molecular tests for spinal muscular atrophy and severe combined immunodeficiency where the programme includes them. The purpose is not diagnosis — it is to flag the infant who needs a confirmatory diagnostic test. [12] [14]
Waisbren and colleagues' study of expanded newborn screening showed that expanded tandem mass spectrometry screening improved outcomes for the disorders it detected without a measurable increase in parental stress, which is part of the evidence base for the expanded panel — but it also produced false positives and mild variants of uncertain significance, which is why every abnormal screen is a call to the confirmatory test, not a diagnosis. The ACMG ACT sheets and confirmatory algorithms specify, for each screened disorder, the immediate action, the confirmatory test, and the clinical follow-up. [13]
The metabolic test panel
The metabolic tests are the diagnostic and the acute-episode tests, not a screen. They are sent in the child with regression, with unexplained encephalopathy, with recurrent unexplained acidosis or hyperammonaemia or hypoglycaemia, with hepatopathy of unknown cause, with cardiomyopathy in the right phenotype, and with a positive newborn bloodspot screen for a metabolic disorder. The principle is the window: send the panel during the decompensation, when the analytes are abnormal, and save aliquots for later; the well-interval panel is often normal and may falsely close the door. [12]
| Test (and sample) | Disorders it flags | Caveat |
|---|---|---|
| Plasma amino acids (heparin or EDTA plasma, on ice) | Phenylketonuria, maple syrup urine disease, the urea-cycle disorders, tyrosinaemia | Interpret against age-specific reference ranges; send during the decompensation (Cite id=12). |
| Urine organic acids (random urine, any urine is better than none) | The organic acidaemias — methylmalonic, propionic, isovaleric; orotic acid in OTC deficiency | Quantitative; the acute-episode sample is the diagnostic one (Cite id=12). |
| Plasma acylcarnitine profile (EDTA plasma) | Fatty-acid oxidation disorders (MCAD, LCHAD, VLCAD); several organic acidaemias | Low free carnitine flags carnitine transporter deficiency; the newborn screen is a subset (Cite id=14). |
| Lactate and pyruvate (venous, free-flowing, on ice) | Mitochondrial disease; pyruvate dehydrogenase and pyruvate carboxylase deficiency | Avoid tourniquet and fist-clenching; a venous sample is an adequate screen (Cite id=12). |
| Transferrin isoforms (serum, isoelectric focusing or MS) | The congenital disorders of glycosylation | Abnormal in CDG types I and II; secondary changes in galactosaemia and fructosaemia (Cite id=12). |
| Very-long-chain fatty acids (serum) | Peroxisomal disorders — X-linked adrenoleukodystrophy, Zellweger spectrum | Elevated in peroxisomal biogenesis defects; send with phytanic acid where indicated (Cite id=12). |
| Ammonia (venous, on ice, to the laboratory within 15 minutes) | Urea-cycle disorders; organic acidaemias; fatty-acid oxidation disorders | A hyperammonaemic infant is a metabolic emergency; treat and sample in parallel (Cite id=12). |
Management — Resuscitation
The immediate priority when a genetic or metabolic test is being considered in a decompensating child is that the treatment precedes the diagnosis. A child in metabolic decompensation — acidotic, hyperammonaemic, hypoglycaemic, or encephalopathic — is resuscitated first, and the diagnostic samples are drawn in parallel before the treatment blunts the abnormality. [12]
A hyperammonaemic infant is the metabolic emergency in which the test and the treatment run together. Ammonia is drawn on ice and run to the laboratory within minutes; the airway, breathing and circulation are secured; an intravenous dextrose-containing fluid is started; protein is stopped; and the detoxification strategy — most commonly nitrogen-scavenger therapy with sodium phenylbutyrate or sodium benzoate, and arginine supplementation — is begun, alongside the search for the cause through plasma amino acids, urine organic acids and the acylcarnitine profile. The threshold for haemofiltration or dialysis in severe hyperammonaemia is decided with the metabolic team and the intensive-care service, because the ammonia must come down before it inflicts irreversible neurological injury. [12]
The critically ill infant with a suspected monogenic disorder is the population for whom rapid whole-genome sequencing is part of the resuscitation. The trio samples are drawn early — EDTA blood from the infant and both parents — so that the sequencing can begin while the intensive-care continues; Meng and colleagues found that an exome diagnosis in the intensive-care unit changed medical management in around a third of diagnosed infants, from withdrawing intensive care to escalating a specific cofactor therapy to redirecting to palliative care. The request is the resuscitation tool. [7] [8]
The abnormal newborn bloodspot screen is a different resuscitation decision — not an emergency, but a time-critical pathway. The infant is recalled, examined, and the confirmatory diagnostic test is sent before the family is told a diagnosis; the ACMG ACT sheet for the flagged disorder specifies the immediate action and the timeframe, and a metabolic specialist or an endocrinologist is engaged within the window the sheet prescribes. A premature or ill infant may have a screen that is falsely abnormal because of physiology, transfusion or parenteral nutrition; the confirmatory test sorts the true from the false. [12]
Management — Definitive & Stepwise
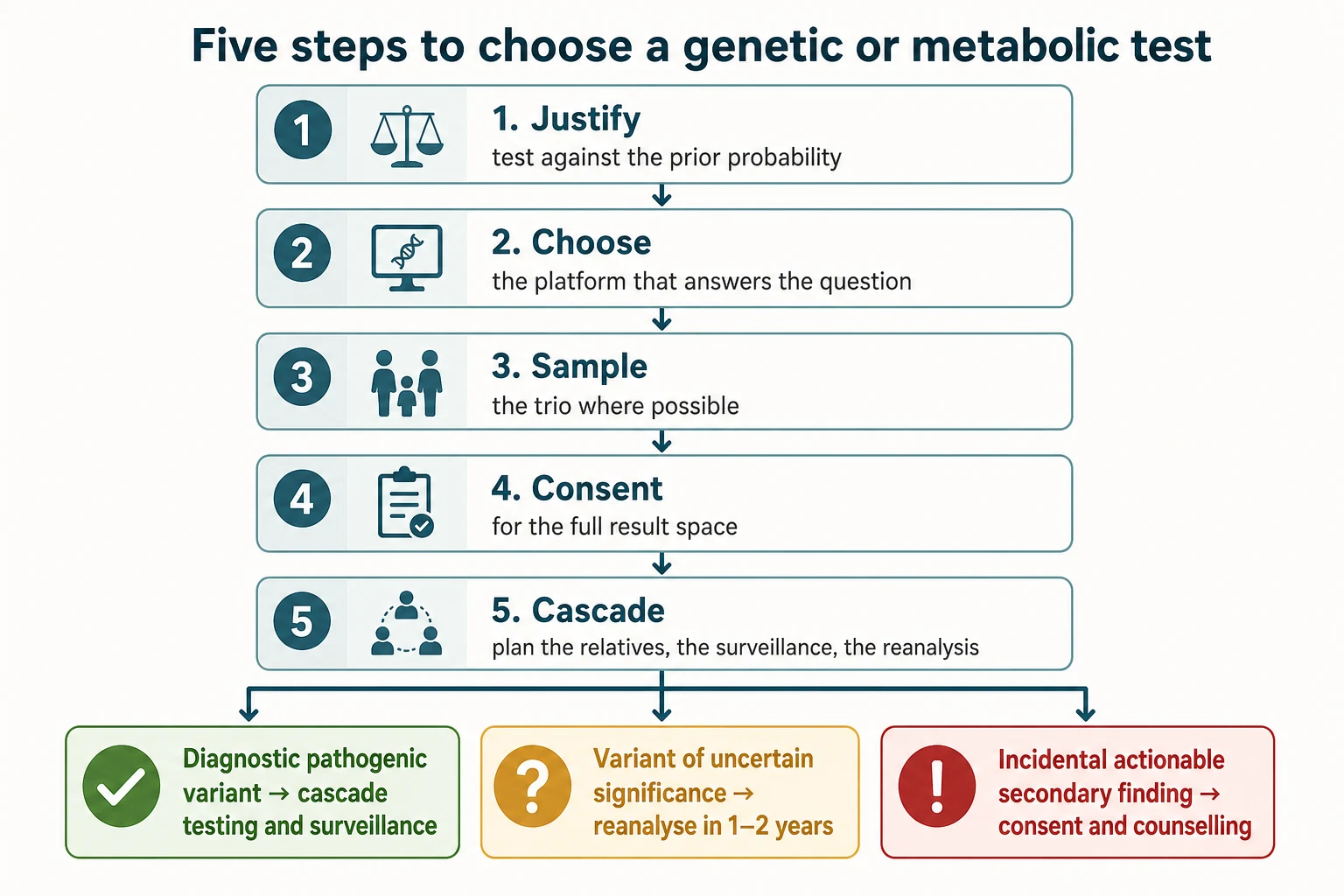
The definitive skill is a stepwise framework that turns the clinical question into the right test, the right sample, the right consent, and the right follow-up. The framework has five steps, and it encodes the family-aware discipline at each one. [1] [10]
Step 1 — Justify the test against the prior probability
Ask whether the test is needed, what question it answers, and what the result will change. A test that will not change management is a test to question. The prior probability shapes the yield — the dysmorphic child with multiple congenital anomalies and developmental delay has a high prior on microarray; the well, non-dysmorphic child with isolated mild speech delay does not. The Miller consensus and the exome series are the worked examples of the high-prior population. [1] [2]
Step 2 — Choose the platform that answers the question
Where the question is a copy-number variant, send the microarray; where the question is a single-nucleotide variant, send the exome or genome; where the question is a metabolic disorder, send the metabolic panel in the right window; where the question is the screened disorder in the newborn, send the confirmatory test the ACT sheet specifies. Do not repeat a normal test — escalate. [1] [2]
Step 3 — Sample for the trio where possible
Trio sampling — the child and both parents — roughly doubles the diagnostic yield of exome over proband-only testing, because the de novo variant is the single most powerful signal in interpretation, and the inherited variants can be filtered by segregation. Request both parents' blood at the same time as the child's, and document the family structure on the request form. Where one parent is unavailable, the extended family and the stored samples may recover some of the yield. [2] [4]
Step 4 — Take consent for the full result space
An exome or genome can return a diagnostic pathogenic variant in a known disease gene, a variant of uncertain significance whose clinical meaning is unresolved, an incidental actionable secondary finding on an ACMG-listed gene (the secondary-findings list stood at 81 genes in the v3.2 update of Miller and colleagues, covering hereditary cancer and cardiovascular risk syndromes among others), or an unexpected finding such as non-paternity or misattributed parentage. The family must have consented to that possibility before the tube is filled, and the consent must record their opt-in or opt-out for secondary findings. [10] [11]
Step 5 — Plan the result communication and the cascade
A molecular diagnosis is a family finding, not a child's finding, and the result communication plans for the cascade testing of at-risk relatives, the reproductive options for the parents (prenatal diagnosis, preimplantation genetic testing in a future pregnancy), the surveillance for the affected child, and the genetic counselling that supports all of it. A negative result is communicated as honestly as a positive one — it does not exclude a genetic cause, and it may turn positive on reanalysis as new genes are described and new variants are reclassified. Plan the reanalysis at one to two years. [6] [11]
Communicating an incidental secondary finding
The communication is the fellowship skill that the viva station examines, and it hinges on the distinction between the diagnostic finding (an answer to the question that prompted the test) and the secondary finding (an answer to a question that was not asked). The ACMG secondary-findings list is the policy that makes some secondary findings reportable by default — the hereditary cancer and cardiovascular risk genes on the list — and the family's opt-out is honoured. The counselling frames the finding as an actionable risk, not a diagnosis; it offers predictive testing to at-risk adult relatives; it does not test minors for adult-onset conditions for which there is no childhood intervention; and it documents the discussion. [10] [11]
Specific Subtypes & Scenarios
Each subtype is the test-selection ladder applied to a specific presentation; the rules above resolve each one. [1] [8]
Global developmental delay and intellectual disability is the worked example of the microarray-first consensus. A four-year-old with unexplained delay is sent a chromosomal microarray as the first-tier test; a normal microarray escalates to whole-exome sequencing as a trio. The yield is around 10 to 20 percent on the microarray and around 25 percent on the exome, and the combined yield is concentrated in the child with dysmorphism, regression or a compatible family history. [1] [2]
Autism spectrum disorder is the population Tammimies and colleagues characterised. The combined yield of microarray and exome is around 16 percent, with the yield concentrated in the child with intellectual disability or dysmorphism; the well, non-dysmorphic child with isolated autism has a lower yield, and that prior shapes whether to test. The first-tier test remains the microarray, escalating to exome where the phenotype concentrates the prior. [5]
Multiple congenital anomalies in the dysmorphic neonate is the population for whom rapid whole-genome sequencing is shifting the paradigm. The microarray is the first-tier test, but in the unstable neonate the rapid trio genome is increasingly first-line, because the diagnostic answer — a recognizable syndrome, a CHARGE or a Noonan or a Kabuki equivalent — directs the cardiac, the renal, the endocrine and the surgical surveillance, and may change the intensive-care trajectory. [1] [8]
The critically ill infant in the NICU with an uninformative septic and metabolic screen is the population the NSIGHT1 trial and the Meng ICU exome series defined. Rapid whole-genome sequencing as a trio yields a diagnosis in around 30 to 40 percent of these infants within days, against around 3 percent with standard testing, and the diagnosis changes management in a meaningful proportion — withdrawing intensive care, escalating a specific therapy, or redirecting to palliation. The platform of choice is the rapid genome, not the sequential single-gene test. [7] [8]
The abnormal newborn bloodspot screen is the pathway the ACMG ACT sheets own. The infant is recalled, examined, and the confirmatory diagnostic test is sent — quantitative plasma amino acids for an elevated phenylalanine, plasma acylcarnitines and a metabolic referral for an elevated octanoylcarnitine, a sweat chloride or CFTR analysis for an elevated immunoreactive trypsinogen, a confirmatory serum 17-hydroxyprogesterone for an elevated screen, confirmatory thyroid function tests for an abnormal thyroxine or thyroid-stimulating hormone, and a molecular confirmation for spinal muscular atrophy or severe combined immunodeficiency. The diagnosis is not made on the screen. [12]
Regression is the presentation that lifts the priority of the metabolic and neurodegenerative workup. The test sequence is metabolic first (urine organic acids, plasma amino acids, acylcarnitines, lactate, very-long-chain fatty acids, transferrin isoforms), brain MRI next, and exome or genome for the child in whom the metabolic and imaging workup is unrevealing. The lysosomal storage disorders, the leukodystrophies, the mitochondrial disorders and the creatine transporter and neurotransmitter disorders are the targets, and the metabolic sample is the acute-episode sample. [12]
The consanguineous family is the population for whom the prior on an autosomal recessive condition is high, and the request should consider an exome or genome rather than sequential targeted tests. The trio sample loses some of its de novo filtering power in a consanguineous family, but the autozygosity mapping and the homozygous-variant analysis recover yield in the recessive architecture. The pre-test counselling includes the discussion of shared parental ancestry and the implications for cascade testing. [4]
Complications & Pitfalls
The complications and pitfalls of genetic and metabolic test selection fall into four categories: the missed diagnosis, the over-tested child, the mishandled result, and the badly taken sample. [1] [12]
The missed diagnosis is the trap of the falsely reassuring test. A normal microarray does not exclude a genetic diagnosis — it tells you to escalate to exome. A normal metabolic panel sent in a well interval does not exclude a metabolic diagnosis — it tells you to re-sample in the next decompensation. A normal exome does not exclude a genetic diagnosis — it tells you to reanalyse in one to two years, or to send a genome for the non-coding and structural variants exome misses. The diagnostic odyssey is the child who is told to come back next year instead of being escalated. [6] [9]
The over-tested child is the trap of the sequential single-gene approach. A child sent for one gene at a time, in series, accumulates cost and delay and consumes the diagnostic sample before the right test is sent. The remedy is the early, well-justified exome or genome, which captures the diagnostic space in a single test and, in the ambulant cohort, lowers the cost per diagnosis. [9]
The mishandled result is the trap of the variant of uncertain significance and the incidental secondary finding. A variant of uncertain significance is reported as uncertain — it is not a diagnosis, it is not a benign finding, and it should not drive surveillance or cascade testing until the evidence firms. An incidental actionable secondary finding is a family finding, and it requires pre-test consent and post-test counselling; testing a minor for an adult-onset condition without a childhood intervention is avoided. The ACMG secondary-findings list and the consent around it are the guardrails. [10] [11]
The badly taken sample is the trap of the uninterpretable result. A newborn bloodspot taken before 48 hours, after a transfusion, or from a squeezed or layered card is uninterpretable and produces false positives and false negatives. A lactate drawn with a tourniquet and a clenched fist is falsely elevated. An ammonia sample not on ice, or delayed to the laboratory, is falsely low. These are sampling errors, owned by the requesting clinician, and they are the reason the bedside sample technique is examined. [12] [14]
The repeat-expansion miss is the trap of the platform's blind spot. Fragile X syndrome, myotonic dystrophy, Friedreich ataxia and the spinocerebellar ataxias are caused by repeat expansions that a microarray cannot detect and that an exome or genome may miss unless the analysis pipeline is configured for repeats. The phenotype drives a targeted repeat-primed polymerase chain reaction or a Southern blot — the exome is not a substitute. [1]
Prognosis & Disposition
The disposition after a genetic or metabolic test follows the result, the child's clinical state, and the family's decisions. [1] [8]
After a diagnostic pathogenic variant, the disposition is the cascade testing of at-risk relatives, the reproductive options for the parents in a future pregnancy (prenatal diagnosis, preimplantation genetic testing), the surveillance for the affected child (the gene-specific surveillance, such as the cardiac surveillance in Noonan and the renal surveillance in tuberous sclerosis), and the genetic counselling that supports all of it. A molecular diagnosis often ends the diagnostic odyssey, but it begins the longitudinal management. [11]
After a variant of uncertain significance, the disposition is the planned reanalysis at one to two years, the segregation analysis in the parents and extended family where it will help, and the honest communication that the result is unresolved. The variant of uncertain significance is not a diagnosis, and it should not drive surveillance or cascade testing until the evidence firms. [6]
After a negative exome or genome, the disposition is the planned reanalysis at one to two years (new genes are described, and variants are reclassified, and the negative test may turn positive on reanalysis), the clinical re-review at intervals, and the return to the clinical picture for a fresh differential. A negative test does not exclude a genetic cause. [6] [9]
After an abnormal newborn bloodspot screen, the disposition is the confirmatory diagnostic test, the engagement of the relevant specialist (metabolic, endocrine, respiratory, immunology, haematology), and the communication to the family once the diagnosis is confirmed. A falsely abnormal screen — common in premature, transfused or parenteral-nutrition infants — is communicated honestly once the confirmatory test sorts the true from the false. [12] [13]
Special Populations
Several populations carry a distinct test-selection trade-off, and the fellowship viva examines the reasoning. [1] [12]
The neonate and young infant is the population for whom rapid whole-genome sequencing is shifting the paradigm in the NICU, and for whom the newborn bloodspot screen is the universal entry point. The trio sample is drawn early, the metabolic panel is sent during the decompensation, and the platform is matched to the acuity — rapid genome for the critically ill, microarray or elective exome for the stable dysmorphic neonate. [7] [8]
The consanguineous family is the population for whom the prior on autosomal recessive disease is high, and the autozygosity mapping of an exome or genome recovers yield in the recessive architecture. The pre-test counselling includes the discussion of shared parental ancestry and the implications for the extended family. [4]
The adopted or migrant child without an informative family history loses the trio advantage and the segregation analysis. The phenotype drives the request, the proband-only exome yields less than the trio, and the absence of a family history does not exclude a genetic cause. Migrant and refugee families may carry population-specific recessive conditions (the haemoglobinopathies, familial Mediterranean fever, specific founder variants), and the test selection respects the population prior. [2]
The child with a life-limiting condition is the population for whom a molecular diagnosis may redirect care from cure to palliation, and the test is offered with the palliative care team and the family at the centre of the decision. The diagnostic answer is valued not for the treatment it enables but for the certainty it provides, the cascade testing it allows, and the reproductive options it opens. [8]
The Indigenous Australian and Māori child is the population for whom equitable access to genomic and metabolic testing is a measurable gap, and for whom the programme delivery — the consent process, the cultural safety of the genetic counselling, the return of results to family and community — is part of the test selection. The request includes the engagement of the Indigenous health liaison and the respect for the family's decision-making structure. [12]
Evidence, Guidelines & Regional Differences
The evidence base for genetic and metabolic test selection rests on a small number of landmark contributions, and the regional guidelines converge on a shared core with local variation. [1] [8]
The Miller 2010 consensus statement is the foundational reference for the microarray-first position, establishing chromosomal microarray as the first-tier clinical diagnostic test for the child with developmental delay, intellectual disability, autism with dysmorphism, or multiple congenital anomalies. It changed practice across paediatrics and informed the United States, United Kingdom, European and Australasian guidelines. [1]
The exome series — Yang and colleagues in the New England Journal of Medicine in 2013, and Lee and colleagues in JAMA in 2014 — established clinical exome sequencing as a diagnostic tool with an overall yield of around 25 percent, and it became the second-tier test after a normal microarray. The Tammimies autism series in JAMA in 2015 quantified the combined yield of microarray and exome in autism at around 16 percent. The Sawyer diagnostic-odyssey series in Clinical Genetics in 2016 named the population-level problem of late and under-sequenced referral. [2] [3] [5] [6]
The rapid-sequencing series — Soden and colleagues in 2014, Meng and colleagues in JAMA Pediatrics in 2017, Tan and colleagues in JAMA Pediatrics in 2017, and the NSIGHT1 randomised controlled trial of Petrikin and colleagues in NPJ Genomic Medicine in 2018 — established that the yield and the management impact of sequencing are highest in the sickest children, and that rapid whole-genome sequencing in the NICU changes management within days. They are the evidence for the rapid paradigm. [4] [7] [8] [9]
The ACMG secondary-findings policy — the 2013 original, the 2016 v2.0 update of Kalia and colleagues, and the 2023 v3.2 list of Miller and colleagues — codified the reportable secondary findings in clinical exome and genome sequencing, and the consent framework that surrounds them. They are the bridge between the technology and the family. [10] [11]
The newborn-screening evidence — Chace and colleagues on tandem mass spectrometry in Clinical Chemistry in 2003, Marsden and colleagues on newborn screening for metabolic disorders in the Journal of Pediatrics in 2006, and Waisbren and colleagues in JAMA in 2003 — established the multianalyte dried-blood-spot screen and its effect on outcomes, with the recognition that the screen is not the diagnosis. The ACMG ACT sheets and confirmatory algorithms give the operational rules for each flagged disorder. [12] [13] [14]
The controversies are real but bounded. The scope of the secondary-findings list — and whether it should expand or contract — is debated, with the ACMG taking the position that the actionable, evidence-based list is reportable by default with opt-out. The role of genomic newborn screening — sequencing every newborn, beyond the screenable conditions — is an active field, with pilot studies and citizens' juries weighing the diagnostic yield against the variant-of-uncertain-significance burden, the data-storage and consent issues, and the equity of access. The reanalysis cadence of a negative exome or genome — and who funds it — is a service-design question that affects every family in the diagnostic odyssey. [6] [11]
Exam Pearls
A candidate who holds these one-liners answers the viva and the single-best-answer question. [1] [8]
- Microarray is the first-tier test for unexplained DD, ID, autism with dysmorphism, and multiple congenital anomalies — the Miller 2010 consensus; yield around 10 to 20 percent; do not repeat once normal.
- A normal microarray does not exclude a genetic diagnosis — it tells you to escalate to exome; the karyotype is reserved for suspected aneuploidy and known familial balanced rearrangements.
- Exome yield is around 25 percent in ambulant children and around 30 to 40 percent in the critically ill infant; trio sampling roughly doubles the yield over proband-only testing.
- Rapid whole-genome sequencing in the NICU changes management in days — the NSIGHT1 RCT yield of around 30 to 40 percent against around 3 percent standard; the platform of choice in the acutely unwell neonate.
- Newborn bloodspot screening is a screen, not a diagnosis — every abnormal result is confirmed by a quantitative diagnostic test before a label is attached; the ACMG ACT sheets give the algorithm.
- The heel-prick is taken at 48 to 72 hours on a Guthrie card, soaked through, never squeezed; earlier or post-transfusion samples are uninterpretable.
- Metabolic tests are sent during the decompensation — plasma amino acids, urine organic acids, acylcarnitines, lactate, ammonia on ice; the well-interval panel is often normal.
- Consent is for the full result space — a diagnostic pathogenic variant, a variant of uncertain significance, an incidental actionable secondary finding on an ACMG-listed gene, or an unexpected non-paternity finding; the secondary-findings list stood at 81 genes in v3.2.
- A negative exome is reanalysed at one to two years — new genes are described and variants are reclassified, and the negative test may turn positive on reanalysis.
References
- [1]Miller DT, Adam MP, Aradhya S, et al Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies Am J Hum Genet, 2010.PMID 20466091
- [2]Yang Y, Muzny DM, Reid JG, et al Clinical whole-exome sequencing for the diagnosis of mendelian disorders N Engl J Med, 2013.PMID 24088041
- [3]Lee H, Deignan JL, Dorrani N, et al Clinical exome sequencing for genetic identification of rare Mendelian disorders JAMA, 2014.PMID 25326637
- [4]Soden SE, Saunders CJ, Willig LK, et al Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders Sci Transl Med, 2014.PMID 25473036
- [5]Tammimies K, Marshall CR, Walker S, et al Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder JAMA, 2015.PMID 26325558
- [6]Sawyer SL, Hartley T, Dyment DA, et al Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care Clin Genet, 2016.PMID 26283276
- [7]Meng L, Pammi M, Saronwala A, et al Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management JAMA Pediatr, 2017.PMID 28973083
- [8]Petrikin JE, Cakici JA, Clark MM, et al The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants NPJ Genom Med, 2018.PMID 29449963
- [9]Tan TY, Dillon OJ, Stark Z, et al Diagnostic Impact and Cost-effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions JAMA Pediatr, 2017.PMID 28759686
- [10]Kalia SS, Adelman K, Bale SJ, et al Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics Genet Med, 2017.PMID 27854360
- [11]Miller DT, Lee K, Abul-Husn NS, et al ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG) Genet Med, 2023.PMID 37347242
- [12]Marsden D, Larson C, Levy HL Newborn screening for metabolic disorders J Pediatr, 2006.PMID 16737864
- [13]Waisbren SE, Albers S, Amato S, et al Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress JAMA, 2003.PMID 14625333
- [14]Chace DH, Kalas TA, Naylor EW Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns Clin Chem, 2003.PMID 14578311